rnalysis.enrichment.RankedSet.single_set_go_enrichment
- RankedSet.single_set_go_enrichment(organism: str | int | Literal['auto'] | Literal['Arabodopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Proteome ID', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', alpha: Fraction = 0.05, min_positive_genes: PositiveInt = 10, lowest_cutoff: Fraction = 0.25, propagate_annotations: Literal['classic', 'elim', 'weight', 'all.m', 'no'] = 'elim', aspects: Literal['any', 'biological_process', 'cellular_component', 'molecular_function'] | Iterable[Literal['biological_process', 'cellular_component', 'molecular_function']] = 'any', evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] | Literal['any'] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', exclude_unannotated_genes: bool = True, return_nonsignificant: bool = False, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', plot_ontology_graph: bool = True, ontology_graph_format: Literal['pdf', 'png', 'svg', 'none'] = 'none', parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) DataFrame | Tuple[DataFrame, Figure]
Calculates enrichment and depletion of the sorted RankedSet for Gene Ontology (GO) terms WITHOUT a background set, using the generalized Minimum Hypergeometric Test (XL-mHG, developed by Prof. Zohar Yakhini and colleagues and generalized by Florian Wagner). The GO terms and annotations are drawn via the GO Solr search engine GOlr, using the search terms defined by the user. P-values are calculated using the generalized Minimum Hypergeometric Test. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment = 0) appears with the smallest value in the scale.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID for which the function will fetch GO annotations.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the GOLR server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
alpha (float between 0 and 1) – Indicates the FDR threshold for significance.
min_positive_genes (a positive int (default=10)) – the minimum number of ‘positive’ genes (genes that match the given attribute) for the enrichment to be considered a valid enrichment. All hypergeometric cutoffs with a smaller number of ‘positive’ genes will not be tested. This is the ‘X’ parameter of the XL-mHG nonparametric test. For example: a value of 10 means that a valid enrichment must have at least 10 ‘positive’ genes to be considered real enrichment.
lowest_cutoff (float between 0 and 1 (default=0.25)) – the lowest cutoff of the hypergeometric that will be tested. This determines the ‘L’ parameter of the XL-mHG nonparametric test. For example: a value of 1 means that every cutoff will be tested. A value of 0.25 means that every cutoff that compares the top 25% or smaller of the list to the rest of the list will be tested.
propagate_annotations ('classic', 'elim', 'weight', 'all.m', or 'no' (default='elim')) – determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/
aspects (str, Iterable of str, 'biological_process', 'molecular_function', 'cellular_component', or 'any' (default='any')) – only annotations from the specified GO aspects will be included in the analysis. Legal aspects are ‘biological_process’ (P), ‘molecular_function’ (F), and ‘cellular_component’ (C).
evidence_types (str, Iterable of str, 'experimental', 'phylogenetic' ,'computational', 'author', 'curator', 'electronic', or 'any' (default='any')) – only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/
excluded_evidence_types (str, Iterable of str, 'experimental', 'phylogenetic' ,'computational', 'author', 'curator', 'electronic', or None (default=None)) – annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/
databases – only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website:
http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :param exclude_unannotated_genes: if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results. :type exclude_unannotated_genes: bool (deafult=True) :param return_nonsignificant: if True, the results DataFrame will include all tested GO terms - both significant and non-significant terms. If False (default), only significant GO terms will be returned. :type return_nonsignificant: bool (default=False) :type save_csv: bool, default False :param save_csv: If True, will save the results to a .csv file, under the name specified in ‘fname’. :type fname: str or pathlib.Path :param fname: The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt. :type return_fig: bool (default=False) :param return_fig: if True, returns a matplotlib Figure object in addition to the results DataFrame. :type plot_horizontal: bool (default=True) :param plot_horizontal: if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot. :param show_expected: if True, the observed/expected values will be shown on the plot. :type show_expected: bool (default=False) :param plot_style: style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set. :type plot_style: ‘bar’ or ‘lollipop’ (default=’bar’) :type plot_ontology_graph: bool (default=True) :param plot_ontology_graph: if True, will generate an ontology graph depicting the significant GO terms and their parent nodes. :type ontology_graph_format: ‘pdf’, ‘png’, ‘svg’, or None (default=None) :param ontology_graph_format: if ontology_graph_format is not ‘none’, the ontology graph will additonally be generated in the specified file format. :type parallel_backend: Literal[PARALLEL_BACKENDS] (default=’loky’) :param parallel_backend: Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise. :rtype: pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure] :return: a pandas DataFrame with the indicated attribute names as rows/index; and a matplotlib Figure, if ‘return_figure’ is set to True.
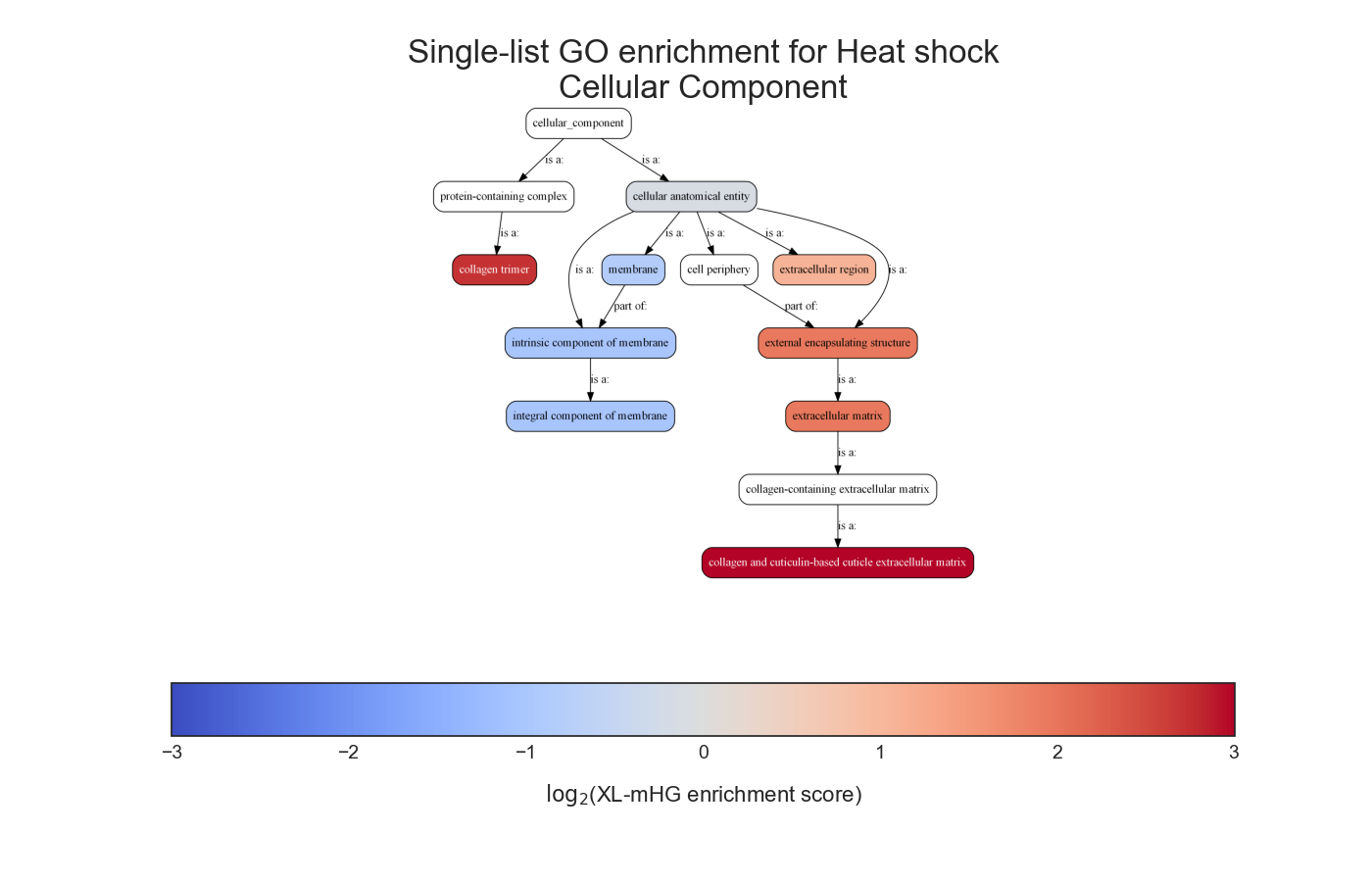
Example plot of single_set_go_enrichment(plot_ontology_graph=True)
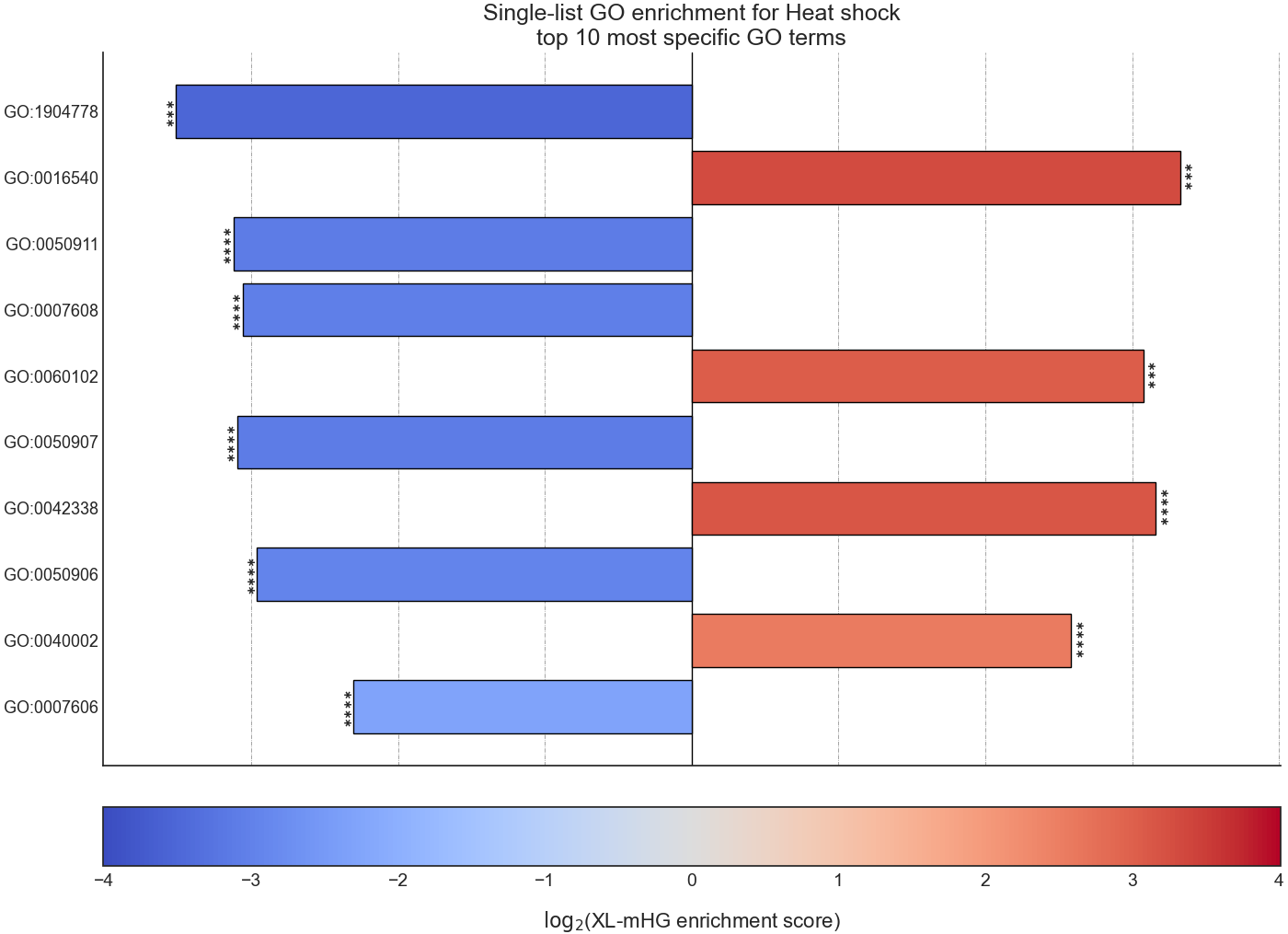
Example plot of single_set_go_enrichment()
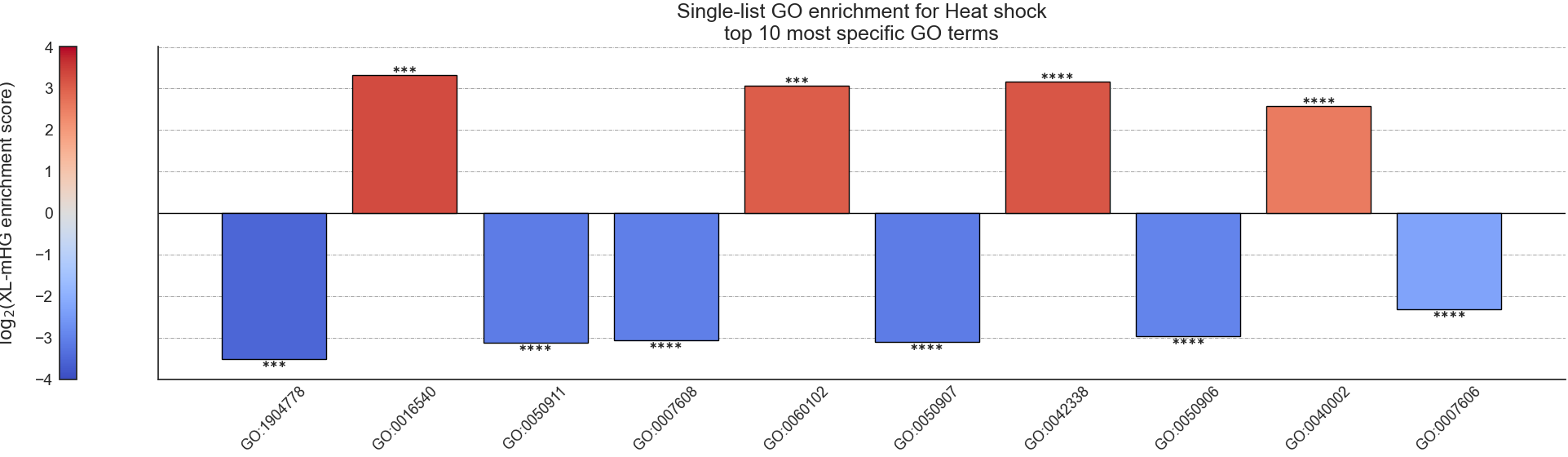
Example plot of single_set_go_enrichment(plot_horizontal = False)