API Reference
Warning
Private functions are not meant to be used out of context, and doing so may lead to unexpected results.
rnalysis.fastq module
The fastq module provides a unified programmatic interface to external tools that process FASTQ files. Those currently include the CutAdapt adapter-trimming tool, the kallisto RNA-sequencing quantification tool, the bowtie2 alignment tool, and the featureCounts feature counting tool.
- class rnalysis.fastq.PairedEndPipeline
Bases:
_FASTQPipeline- _func_signature(func: LambdaType, args: tuple, kwargs: dict)
Returns a string functions signature for the given function and arguments.
- Parameters:
func (function) – the function or method to generate signature for
args (tuple) – arguments given for the function
kwargs (dict) – keyworded arguments given for the function
- Returns:
function signature string
- Return type:
str
- static _param_string(args: tuple, kwargs: dict)
Returns a formatted string of the given arguments and keyworded arguments.
- Parameters:
args (tuple) – arguments to format as string
kwargs (dict) – keyworded arguments to format as string
- Returns:
a formatted string of arguments and keyworded argumentss
- Return type:
str
- _readable_func_signature(func: LambdaType, args: tuple, kwargs: dict)
Returns a human-readable string functions signature for the given function and arguments.
- Parameters:
func (function) – the function or method to generate signature for
args (tuple) – arguments given for the function
kwargs (dict) – keyworded arguments given for the function
- Returns:
function signature string
- Return type:
str
- export_pipeline(filename: str | Path | None) None | str
Export a Pipeline to a Pipeline YAML file or YAML-like string.
- Parameters:
filename (str, pathlib.Path, or None) – filename to save the Pipeline YAML to, or None to return a YAML-like string instead.
- Returns:
if filename is None, returns the Pipeline YAML-like string.
- functions
list of functions to perform
- classmethod import_pipeline(filename: str | Path) GenericPipeline
Import a Pipeline from a Pipeline YAML file or YAML-like string.
- Parameters:
filename (str or pathlib.Path) – name of the YAML file containing the Pipeline, or a YAML-like string.
- Returns:
the imported Pipeline
- Return type:
- params
list of function parameters
- remove_last_function()
Removes from the Pipeline the last function that was added to it. Removal is in-place.
- Examples:
>>> from rnalysis import filtering >>> pipe = filtering.Pipeline() >>> pipe.add_function(filtering.Filter.filter_missing_values) Added function 'Filter.filter_missing_values()' to the pipeline. >>> pipe.remove_last_function() Removed function filter_missing_values with parameters [] from the pipeline.
- class rnalysis.fastq.SingleEndPipeline
Bases:
_FASTQPipeline- _func_signature(func: LambdaType, args: tuple, kwargs: dict)
Returns a string functions signature for the given function and arguments.
- Parameters:
func (function) – the function or method to generate signature for
args (tuple) – arguments given for the function
kwargs (dict) – keyworded arguments given for the function
- Returns:
function signature string
- Return type:
str
- static _param_string(args: tuple, kwargs: dict)
Returns a formatted string of the given arguments and keyworded arguments.
- Parameters:
args (tuple) – arguments to format as string
kwargs (dict) – keyworded arguments to format as string
- Returns:
a formatted string of arguments and keyworded argumentss
- Return type:
str
- _readable_func_signature(func: LambdaType, args: tuple, kwargs: dict)
Returns a human-readable string functions signature for the given function and arguments.
- Parameters:
func (function) – the function or method to generate signature for
args (tuple) – arguments given for the function
kwargs (dict) – keyworded arguments given for the function
- Returns:
function signature string
- Return type:
str
- export_pipeline(filename: str | Path | None) None | str
Export a Pipeline to a Pipeline YAML file or YAML-like string.
- Parameters:
filename (str, pathlib.Path, or None) – filename to save the Pipeline YAML to, or None to return a YAML-like string instead.
- Returns:
if filename is None, returns the Pipeline YAML-like string.
- functions
list of functions to perform
- classmethod import_pipeline(filename: str | Path) GenericPipeline
Import a Pipeline from a Pipeline YAML file or YAML-like string.
- Parameters:
filename (str or pathlib.Path) – name of the YAML file containing the Pipeline, or a YAML-like string.
- Returns:
the imported Pipeline
- Return type:
- params
list of function parameters
- remove_last_function()
Removes from the Pipeline the last function that was added to it. Removal is in-place.
- Examples:
>>> from rnalysis import filtering >>> pipe = filtering.Pipeline() >>> pipe.add_function(filtering.Filter.filter_missing_values) Added function 'Filter.filter_missing_values()' to the pipeline. >>> pipe.remove_last_function() Removed function filter_missing_values with parameters [] from the pipeline.
- class rnalysis.fastq._FASTQPipeline
Bases:
GenericPipeline,ABC- _func_signature(func: LambdaType, args: tuple, kwargs: dict)
Returns a string functions signature for the given function and arguments.
- Parameters:
func (function) – the function or method to generate signature for
args (tuple) – arguments given for the function
kwargs (dict) – keyworded arguments given for the function
- Returns:
function signature string
- Return type:
str
- static _param_string(args: tuple, kwargs: dict)
Returns a formatted string of the given arguments and keyworded arguments.
- Parameters:
args (tuple) – arguments to format as string
kwargs (dict) – keyworded arguments to format as string
- Returns:
a formatted string of arguments and keyworded argumentss
- Return type:
str
- _readable_func_signature(func: LambdaType, args: tuple, kwargs: dict)
Returns a human-readable string functions signature for the given function and arguments.
- Parameters:
func (function) – the function or method to generate signature for
args (tuple) – arguments given for the function
kwargs (dict) – keyworded arguments given for the function
- Returns:
function signature string
- Return type:
str
- export_pipeline(filename: str | Path | None) None | str
Export a Pipeline to a Pipeline YAML file or YAML-like string.
- Parameters:
filename (str, pathlib.Path, or None) – filename to save the Pipeline YAML to, or None to return a YAML-like string instead.
- Returns:
if filename is None, returns the Pipeline YAML-like string.
- functions
list of functions to perform
- classmethod import_pipeline(filename: str | Path) GenericPipeline
Import a Pipeline from a Pipeline YAML file or YAML-like string.
- Parameters:
filename (str or pathlib.Path) – name of the YAML file containing the Pipeline, or a YAML-like string.
- Returns:
the imported Pipeline
- Return type:
- params
list of function parameters
- remove_last_function()
Removes from the Pipeline the last function that was added to it. Removal is in-place.
- Examples:
>>> from rnalysis import filtering >>> pipe = filtering.Pipeline() >>> pipe.add_function(filtering.Filter.filter_missing_values) Added function 'Filter.filter_missing_values()' to the pipeline. >>> pipe.remove_last_function() Removed function filter_missing_values with parameters [] from the pipeline.
- rnalysis.fastq._merge_kallisto_outputs(output_folder: str | Path, new_sample_names: List[str])
output a merged csv file of transcript estimated counts, and a merged csv file of transcript estimated TPMs.
- rnalysis.fastq.bowtie2_align_paired_end(r1_files: List[str], r2_files: List[str], output_folder: str | Path, index_file: str | Path, bowtie2_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto', 'smart'] = 'smart', mode: Literal['end-to-end', 'local'] = 'end-to-end', settings_preset: Literal['very-fast', 'fast', 'sensitive', 'very-sensitive'] = 'very-sensitive', ignore_qualities: bool = False, quality_score_type: Literal['phred33', 'phred64', 'solexa-quals', 'int-quals'] = 'phred33', mate_orientations: Literal['fwd-rev', 'rev-fwd', 'fwd-fwd'] = 'fwd-rev', min_fragment_length: NonNegativeInt = 0, max_fragment_length: PositiveInt = 500, allow_individual_alignment: bool = True, allow_disconcordant_alignment: bool = True, random_seed: NonNegativeInt = 0, threads: PositiveInt = 1)
Align paired-end reads from FASTQ files to a reference sequence using the bowtie2 aligner. The FASTQ file pairs will be individually aligned, and the aligned SAM files will be saved in the output folder. You can read more about how bowtie2 works in the bowtie2 manual.
- Parameters:
r1_files (list of str/Path to existing FASTQ files) – a list of paths to your Read#1 files. The files should be sorted in tandem with r2_files, so that they line up to form pairs of R1 and R2 files.
r2_files (list of str/Path to existing FASTQ files) – a list of paths to your Read#2 files. The files should be sorted in tandem with r1_files, so that they line up to form pairs of R1 and R2 files.
output_folder (str/Path to an existing folder) – Path to a folder in which the aligned reads, as well as the log files, will be saved.
index_file (str or Path) – Path to a pre-built bowtie2 index of the target genome. Can either be downloaded from the bowtie2 website (menu on the right), or generated manually from FASTA files using the function ‘bowtie2_create_index’. Note that bowtie2 indices are composed of multiple files ending with the ‘.bt2’ suffix. All of those files should be in the same location. It is enough to specify the path to one of those files (for example, ‘path/to/index.1.bt2’), or to the main name of the index (for example, ‘path/to/index’).
bowtie2_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of bowtie2. For example: ‘C:/Program Files/bowtie2-2.5.1’. if installation folder is set to ‘auto’, RNAlysis will attempt to find it automatically.
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each quantified sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the file pairs.
mode ('end-to-end' or 'local' (default='end-to-end')) – determines the alignment mode of bowtie2. end-to-end mode will look for alignments involving all the read characters. local mode will allow ‘clipping’ of nucleotides from both sides of the read, if that maximizes the alignment score.
settings_preset ('very-sensitive', 'sensitive', 'fast', or 'very-fast' (default='very-sensitive')) – determines the alignment sensitivity preset. Higher sensitivity will result in more accurate alignments, but will take longer to calculate. You can read more about the settings presets in the bowtie2 manual.
ignore_qualities (bool (default=False)) – if True, bowtie2 will ignore the qualities of the reads and treat them all as maximum quality.
quality_score_type ('phred33', 'phred64', 'solexa-quals', or 'int-quals' (default='phred33')) – determines the encoding type of the read quality scores. Most modern sequencing setups use phred+33.
mate_orientations ('fwd-rev', 'rev-fwd', or 'fwd-fwd' (default='fwd-rev'))
min_fragment_length (int >= 0 (default=0)) – The minimum fragment length for valid paired-end alignments.
max_fragment_length (int > 0 (default=500)) – The maximum fragment length for valid paired-end alignments.
allow_individual_alignment (bool (default=)
allow_disconcordant_alignment (bool (default=)
random_seed (int >=0 (default=0)) – determines the seed for pseudo-random number generator.
threads (int > 0 (default=1)) – number of threads to run bowtie2-build on. More threads will generally make index building faster.
- rnalysis.fastq.bowtie2_align_single_end(fastq_folder: str | Path, output_folder: str | Path, index_file: str | Path, bowtie2_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', mode: Literal['end-to-end', 'local'] = 'end-to-end', settings_preset: Literal['very-fast', 'fast', 'sensitive', 'very-sensitive'] = 'very-sensitive', ignore_qualities: bool = False, quality_score_type: Literal['phred33', 'phred64', 'solexa-quals', 'int-quals'] = 'phred33', random_seed: NonNegativeInt = 0, threads: PositiveInt = 1)
Align single-end reads from FASTQ files to a reference sequence using the bowtie2 aligner. The FASTQ files will be individually aligned, and the aligned SAM files will be saved in the output folder. You can read more about how bowtie2 works in the bowtie2 manual.
- Parameters:
fastq_folder (str or Path) – Path to the folder containing the FASTQ files you want to quantify
output_folder (str/Path to an existing folder) – Path to a folder in which the aligned reads, as well as the log files, will be saved.
index_file (str or Path) –
Path to a pre-built bowtie2 index of the target genome. Can either be downloaded from the bowtie2 website (menu on the right), or generated manually from FASTA files using the function ‘bowtie2_create_index’. Note that bowtie2 indices are composed of multiple files ending with the ‘.bt2’ suffix. All of those files should be in the same location. It is enough to specify the path to one of those files (for example, ‘path/to/index.1.bt2’), or to the main name of the index (for example, ‘path/to/index’).
bowtie2_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of bowtie2. For example: ‘C:/Program Files/bowtie2-2.5.1’. if installation folder is set to ‘auto’, RNAlysis will attempt to find it automatically.
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each quantified sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the file pairs.
mode ('end-to-end' or 'local' (default='end-to-end')) – determines the alignment mode of bowtie2. end-to-end mode will look for alignments involving all the read characters. local mode will allow ‘clipping’ of nucleotides from both sides of the read, if that maximizes the alignment score.
settings_preset ('very-sensitive', 'sensitive', 'fast', or 'very-fast' (default='very-sensitive')) – determines the alignment sensitivity preset. Higher sensitivity will result in more accurate alignments, but will take longer to calculate. You can read more about the settings presets in the bowtie2 manual.
ignore_qualities (bool (default=False)) – if True, bowtie2 will ignore the qualities of the reads and treat them all as maximum quality.
quality_score_type ('phred33', 'phred64', 'solexa-quals', or 'int-quals' (default='phred33')) – determines the encoding type of the read quality scores. Most modern sequencing setups use phred+33.
random_seed (int >=0 (default=0)) – determines the seed for pseudo-random number generator.
threads (int > 0 (default=1)) – number of threads to run bowtie2-build on. More threads will generally make index building faster.
- rnalysis.fastq.bowtie2_create_index(genome_fastas: List[str | Path], output_folder: str | Path, index_name: str | Literal['auto'] = 'auto', bowtie2_installation_folder: str | Path | Literal['auto'] = 'auto', random_seed: NonNegativeInt | None = None, threads: PositiveInt = 1)
builds a bowtie index from FASTA formatted files of target sequences (genome). The index files will be saved in the same folder as your first FASTA file, with the .bt2 suffix. Be aware that there are pre-built bowtie2 indices for popular model organisms. These can be downloaded from the bowtie2 website (from menu on the right).
- Parameters:
genome_fastas (list of str or Path) – Path to the FASTA file/files which contain reference sequences to be aligned to.
output_folder (str or Path) – Path to the folder in which the bowtie2 index files will be saved.
index_name (str or 'auto' (default='auto')) – The basename of the index files. bowtie2 will create files named index_name.1.bt2, index_name.2.bt2, index_name.3.bt2, index_name.4.bt2, index_name.rev.1.bt2, and index_name.rev.2.bt2. if index_name=’auto’, the index name used will be the stem of the first supplied genome FASTA file (for example: if the first genome FASTA file is ‘path/to/genome.fa.gz’, the index name will be ‘genome’).
bowtie2_installation_folder – Path to the installation folder of bowtie2. For example:
‘C:/Program Files/bowtie2-2.5.1’. if installation folder is set to ‘auto’, RNAlysis will attempt to find it automatically. :type bowtie2_installation_folder: str, Path, or ‘auto’ (default=’auto’) :param random_seed: if specified, determines the seed for pseudo-random number generator. :type random_seed: int >=0 or None (default=None) :param threads: number of threads to run bowtie2-build on. More threads will generally make index building faster. :type threads: int > 0 (default=1)
- rnalysis.fastq.convert_sam_format(input_folder: str | Path, output_folder: str | Path, picard_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', output_format: Literal['sam', 'bam'] = 'bam')
Convert SAM files to BAM files or vice versa using Picard SamFormatConverter.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to convert.
output_folder (str or Path) – Path to a folder in which the converted FASTQ files will be saved.
picard_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of Picard. For example: ‘C:/Program Files/Picard’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each converted sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the files in the directory.
output_format ('sam' or 'bam' (default='bam')) – format to convert the files into.
- rnalysis.fastq.fastq_to_sam_paired(r1_files: List[str], r2_files: List[str], output_folder: str | Path, picard_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', output_format: Literal['sam', 'bam'] = 'bam', quality_score_type: Literal['auto'] | Literal['phred33', 'phred64', 'solexa-quals', 'int-quals'] = 'auto')
Convert SAM/BAM files to FASTQ files using Picard SamToFastq.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to convert.
output_folder (str or Path) – Path to a folder in which the converted FASTQ files will be saved.
picard_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of Picard. For example: ‘C:/Program Files/Picard’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each converted sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the files in the directory.
- Returns:
a list of the paths to the generated FASTQ files.
- Return type:
list of str
- rnalysis.fastq.fastq_to_sam_single(input_folder: str | Path, output_folder: str | Path, picard_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', output_format: Literal['sam', 'bam'] = 'bam', quality_score_type: Literal['auto'] | Literal['phred33', 'phred64', 'solexa-quals', 'int-quals'] = 'auto')
Convert SAM/BAM files to FASTQ files using Picard SamToFastq.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to convert.
output_folder (str or Path) – Path to a folder in which the converted FASTQ files will be saved.
picard_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of Picard. For example: ‘C:/Program Files/Picard’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each converted sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the files in the directory.
- Returns:
a list of the paths to the generated FASTQ files.
- Return type:
list of str
- rnalysis.fastq.featurecounts_paired_end(input_folder: str | Path, output_folder: str | Path, gtf_file: str | Path, gtf_feature_type: str = 'exon', gtf_attr_name: str = 'gene_id', r_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', stranded: Literal['no', 'forward', 'reverse'] = 'no', min_mapping_quality: int = 0, count_multi_mapping_reads: bool = False, count_multi_overlapping_reads: bool = False, ignore_secondary: bool = True, count_fractionally: bool = False, is_long_read: bool = False, require_both_mapped: bool = True, count_chimeric_fragments: bool = False, min_fragment_length: NonNegativeInt = 50, max_fragment_length: PositiveInt | None = 600, report_read_assignment: Literal['bam', 'sam', 'core'] | None = None, threads: PositiveInt = 1, return_log: bool = False) Tuple[CountFilter, DataFrame, DataFrame] | Tuple[CountFilter, DataFrame, DataFrame, Path]
Assign mapped paired-end sequencing reads to specified genomic features using RSubread featureCounts. Returns a count matrix (CountFilter) containing feature counts for all input files, a DataFrame summarizing the features reads were aligned to, and a DataFrame summarizing the alignment statistics.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to quantfy.
output_folder (str or Path) – Path to a folder in which the quantified results, as well as the log files and R script used to generate them, will be saved.
gtf_file (str or Path) – Path to a GTF annotation file. This file will be used to map reads to features. The chromosome names in the GTF files should match the ones in the index file with which you aligned the reads.
gtf_feature_type (str (default='exon')) – the feature type or types used to select rows in the GTF annotation which will be used for read summarization.
gtf_attr_name (str (default='gene_id')) – the attribute type in the GTF annotation which will be used to group features (eg. exons) into meta-features (eg. genes).
r_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of R. For example: ‘C:/Program Files/R/R-4.2.1’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each quantified sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the file pairs.
stranded ('no', 'forward', 'reverse' (default='no')) – Indicates the strandedness of the data. ‘no’ indicates the data is not stranded. ‘forward’ indicates the data is stranded, where the first read in the pair aligns to the forward strand of a transcript. ‘reverse’ indicates the data is stranded, where the first read in the pair aligns to the reverse strand of a transcript.
min_mapping_quality (int >= 0 (default=0)) – the minimum mapping quality score a read must satisfy in order to be counted. For paired-end reads, at least one end should satisfy this criteria.
count_multi_mapping_reads (bool (default=True)) – indicating if multi-mapping reads/fragments should be counted (‘NH’ tag in BAM/SAM files).
count_multi_overlapping_reads (bool (default=False)) – indicating if a read is allowed to be assigned to more than one feature (or meta-feature) if it is found to overlap with more than one feature (or meta-feature).
ignore_secondary (bool (default=True)) – indicating if only primary alignments should be counted. Primary and secondary alignments are identified using bit 0x100 in the Flag field of SAM/BAM files. If True, all primary alignments in a dataset will be counted no matter they are from multi-mapping reads or not.
count_fractionally (bool (default=False)) – indicating if fractional counts are produced for multi-mapping reads and/or multi-overlapping reads.
is_long_read (bool (default=False)) – indicating if input data contain long reads. This option should be set to True if counting Nanopore or PacBio long reads.
report_read_assignment ('bam', 'sam', 'core', or None (default=None)) – if not None, featureCounts will generated detailed read assignment results for each read pair. These results can be saved in one of three formats: BAM, SAM, or CORE.
require_both_mapped (bool (default=True)) – indicating if both ends from the same fragment are required to be successfully aligned before the fragment can be assigned to a feature or meta-feature.
count_chimeric_fragments (bool(default=False)) – indicating whether a chimeric fragment, which has its two reads mapped to different chromosomes, should be counted or not.
min_fragment_length (int >= 0 (default=50)) – The minimum fragment length for valid paired-end alignments. Read pairs with shorter fragments will not be counted.
max_fragment_length (int > 0 or None (default=600)) – The maximum fragment length for valid paired-end alignments. Read pairs with longer fragments will not be counted.
threads (int > 0 (default=1)) – number of threads to run bowtie2-build on. More threads will generally make index building faster.
return_log (bool (default=False)) – if True, the function will return the path to the analysis logfile, which includes session info.
- Returns:
a count matrix (CountFilter) containing feature counts for all input files, a DataFrame summarizing the features reads were aligned to, and a DataFrame summarizing the alignment statistics.
- Return type:
(filtering.CountFilter, pl.DataFrame, pl.DataFrame)
- rnalysis.fastq.featurecounts_single_end(input_folder: str | Path, output_folder: str | Path, gtf_file: str | Path, gtf_feature_type: str = 'exon', gtf_attr_name: str = 'gene_id', r_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', stranded: Literal['no', 'forward', 'reverse'] = 'no', min_mapping_quality: int = 0, count_multi_mapping_reads: bool = False, count_multi_overlapping_reads: bool = False, ignore_secondary: bool = True, count_fractionally: bool = False, is_long_read: bool = False, report_read_assignment: Literal['bam', 'sam', 'core'] | None = None, threads: PositiveInt = 1, return_log: bool = False) Tuple[CountFilter, DataFrame, DataFrame] | Tuple[CountFilter, DataFrame, DataFrame, Path]
Assign mapped single-end sequencing reads to specified genomic features using RSubread featureCounts. Returns a count matrix (CountFilter) containing feature counts for all input files, a DataFrame summarizing the features reads were aligned to, and a DataFrame summarizing the alignment statistics.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to quantfy.
output_folder (str or Path) – Path to a folder in which the quantified results, as well as the log files, will be saved.
gtf_file (str or Path) – Path to a GTF annotation file. This file will be used to map reads to features. The chromosome names in the GTF files should match the ones in the index file with which you aligned the reads.
gtf_feature_type (str (default='exon')) – the feature type or types used to select rows in the GTF annotation which will be used for read summarization.
gtf_attr_name (str (default='gene_id')) – the attribute type in the GTF annotation which will be used to group features (eg. exons) into meta-features (eg. genes).
r_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of R. For example: ‘C:/Program Files/R/R-4.2.1’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each quantified sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the alphabetical order of the files in the directory.
stranded ('no', 'forward', 'reverse' (default='no')) – Indicates the strandedness of the data. ‘no’ indicates the data is not stranded. ‘forward’ indicates the data is stranded, where the reads align to the forward strand of a transcript. ‘reverse’ indicates the data is stranded, where the reads align to the reverse strand of a transcript.
min_mapping_quality (int >= 0 (default=0)) – the minimum mapping quality score a read must satisfy in order to be counted.
count_multi_mapping_reads (bool (default=True)) – indicating if multi-mapping reads/fragments should be counted (‘NH’ tag in BAM/SAM files).
count_multi_overlapping_reads (bool (default=False)) – indicating if a read is allowed to be assigned to more than one feature (or meta-feature) if it is found to overlap with more than one feature (or meta-feature).
ignore_secondary (bool (default=True)) – indicating if only primary alignments should be counted. Primary and secondary alignments are identified using bit 0x100 in the Flag field of SAM/BAM files. If True, all primary alignments in a dataset will be counted no matter they are from multi-mapping reads or not.
count_fractionally (bool (default=False)) – indicating if fractional counts are produced for multi-mapping reads and/or multi-overlapping reads.
is_long_read (bool (default=False)) – indicating if input data contain long reads. This option should be set to True if counting Nanopore or PacBio long reads.
report_read_assignment ('bam', 'sam', 'core', or None (default=None)) – if not None, featureCounts will generated detailed read assignment results for each read. These results can be saved in one of three formats: BAM, SAM, or CORE.
threads (int > 0 (default=1)) – number of threads to run bowtie2-build on. More threads will generally make index building faster.
return_log (bool (default=False)) – if True, the function will return the path to the analysis logfile, which includes session info.
- Returns:
a count matrix (CountFilter) containing feature counts for all input files, a DataFrame summarizing the features reads were aligned to, and a DataFrame summarizing the alignment statistics.
- Return type:
(filtering.CountFilter, pl.DataFrame, pl.DataFrame)
- rnalysis.fastq.find_duplicates(input_folder: str | Path, output_folder: str | Path, picard_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', output_format: Literal['sam', 'bam'] = 'bam', duplicate_handling: Literal['mark', 'remove_optical', 'remove_all'] = 'remove_all', duplicate_scoring_strategy: Literal['reference_length', 'sum_of_base_qualities', 'random'] = 'sum_of_base_qualities', optical_duplicate_pixel_distance: int = 100)
Find duplicate reads in SAM/BAM files using Picard MarkDuplicates.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to sort.
output_folder (str or Path) – Path to a folder in which the sorted SAM/BAM files will be saved.
picard_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of Picard. For example: ‘C:/Program Files/Picard’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each converted sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the files in the directory.
output_format ('sam' or 'bam' (default='bam')) – Format of the output file.
duplicate_handling ('mark', 'remove_optical', or 'remove_all' (default='remove_all')) – How to handle detected duplicate reads. If ‘mark’, duplicate reads will be marked with a 1024 flag. If ‘remove_optical’, ‘optical’ duplicates and other duplicates that appear to have arisen from the sequencing process instead of the library preparation process will be removed. If ‘remove_all’, all duplicate reads will be removed.
duplicate_scoring_strategy ('reference_length', 'sum_of_base_qualities', or 'random' (default='sum_of_base_qualities')) – How to score duplicate reads. If ‘reference_length’, the length of the reference sequence will be used. If ‘sum_of_base_qualities’, the sum of the base qualities will be used.
optical_duplicate_pixel_distance (int (default=100)) – The maximum offset between two duplicate clusters in order to consider them optical duplicates. The default (100) is appropriate for unpatterned versions of the Illumina platform. For the patterned flowcell models, 2500 is moreappropriate. For other platforms and models, users should experiment to find what works best.
- rnalysis.fastq.kallisto_create_index(transcriptome_fasta: str | Path, kallisto_installation_folder: str | Path | Literal['auto'] = 'auto', kmer_length: PositiveInt = 31, make_unique: bool = False)
builds a kallisto index from a FASTA formatted file of target sequences (transcriptome). The index file will be saved in the same folder as your FASTA file, with the .idx suffix. Be aware that there are pre-built kallisto indices for popular model organisms. These can be downloaded from the kallisto transcriptome indices site.
- Parameters:
transcriptome_fasta (str or Path) – Path to the FASTA file of your desired transcriptome.
kallisto_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of kallisto. For example: ‘C:/Program Files/kallisto’. if installation folder is set to ‘auto’, RNAlysis will attempt to find it automatically.
kmer_length (an odd int between 1 and 31 (default=31)) – k-mer length of the index.
make_unique (bool (default=False)) – if True, replace repeated target names with unique names.
- rnalysis.fastq.kallisto_quantify_paired_end(r1_files: List[str], r2_files: List[str], output_folder: str | Path, index_file: str | Path, gtf_file: str | Path, kallisto_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto', 'smart'] = 'smart', stranded: Literal['no', 'forward', 'reverse'] = 'no', summation_method: Literal['scaled_tpm', 'raw'] = 'scaled_tpm', bootstrap_samples: PositiveInt | None = None, **legacy_args) CountFilter
Quantify transcript abundance in paired-end mRNA sequencing data using kallisto. The FASTQ file pairs will be individually quantified and saved in the output folder, each in its own sub-folder. Alongside these files, three .csv files will be saved: a per-transcript count estimate table, a per-transcript TPM estimate table, and a per-gene scaled output table. The per-gene scaled output table is generated using the scaledTPM method (scaling the TPM estimates up to the library size) as described by Soneson et al 2015 and used in the tximport R package. This table format is considered un-normalized for library size, and can therefore be used directly by count-based statistical inference tools such as DESeq2. RNAlysis will return this table once the analysis is finished.
- Parameters:
summation_method ('scaled_tpm' or 'raw' (default='scaled_tpm'))
r1_files (list of str/Path to existing FASTQ files) – a list of paths to your Read#1 files. The files should be sorted in tandem with r2_files, so that they line up to form pairs of R1 and R2 files.
r2_files (list of str/Path to existing FASTQ files) – a list of paths to your Read#2 files. The files should be sorted in tandem with r1_files, so that they line up to form pairs of R1 and R2 files.
output_folder (str/Path to an existing folder) – Path to a folder in which the quantified results, as well as the log files, will be saved. The individual output of each pair of FASTQ files will reside in a different sub-folder within the output folder, and a summarized results table will be saved in the output folder itself.
index_file (str or Path) –
Path to a pre-built kallisto index of the target transcriptome. Can either be downloaded from the kallisto transcriptome indices site, or generated manually from a FASTA file using the function kallisto_create_index.
gtf_file (str or Path) – Path to a GTF annotation file. This file will be used to map per-transcript abundances to per-gene estimated counts. The transcript names in the GTF files should match the ones in the index file - we recommend downloading cDNA FASTA/index files and GTF files from the same data source.
kallisto_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of kallisto. For example: ‘C:/Program Files/kallisto’. if installation folder is set to ‘auto’, RNAlysis will attempt to find it automatically.
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each quantified sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the file pairs.
stranded ('no', 'forward', 'reverse' (default='no')) – Indicates the strandedness of the data. ‘no’ indicates the data is not stranded. ‘forward’ indicates the data is stranded, where the first read in the pair pseudoaligns to the forward strand of a transcript. ‘reverse’ indicates the data is stranded, where the first read in the pair pseudoaligns to the reverse strand of a transcript.
summation_method – Determines the method used to sum the transcript-level abundances to gene-level abundances. ‘scaled_tpm’ sums the transcript TPM estimates the gene level, and then scales then to the library size. ‘raw’ sums the transcript estimated counts to the gene level without scaling.
learn_bias (bool (default=False)) – if True, kallisto learns parameters for a model of sequences specific bias and corrects the abundances accordlingly. Note that this feature is not supported by kallisto versions beyond 0.48.0.
seek_fusion_genes (bool (default=False)) – if True, does normal quantification, but additionally looks for reads that do not pseudoalign because they are potentially from fusion genes. All output is written to the file fusion.txt in the output folder. Note that this feature is not supported by kallisto versions beyond 0.48.0.
bootstrap_samples (int >0 or None (default=None)) – Number of bootstrap samples to be generated. Bootstrap samples do not affect the estimated count values, but generates an additional .hdf5 output file which contains uncertainty estimates for the expression levels. This is required if you use tools such as Sleuth for downstream differential expression analysis, but not for more traditional tools such as DESeq2 and edgeR.
- rnalysis.fastq.kallisto_quantify_single_end(fastq_folder: str | Path, output_folder: str | Path, index_file: str | Path, gtf_file: str | Path, average_fragment_length: float, stdev_fragment_length: float, kallisto_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', stranded: Literal['no', 'forward', 'reverse'] = 'no', summation_method: Literal['scaled_tpm', 'raw'] = 'scaled_tpm', bootstrap_samples: PositiveInt | None = None, **legacy_args) CountFilter
Quantify transcript abundance in single-end mRNA sequencing data using kallisto. The FASTQ files will be individually quantified and saved in the output folder, each in its own sub-folder. Alongside these files, three .csv files will be saved: a per-transcript count estimate table, a per-transcript TPM estimate table, and a per-gene scaled output table. The per-gene scaled output table is generated using the scaledTPM method (scaling the TPM estimates up to the library size) as described by Soneson et al 2015 and used in the tximport R package. This table format is considered un-normalized for library size, and can therefore be used directly by count-based statistical inference tools such as DESeq2. RNAlysis will return this table once the analysis is finished.
- Parameters:
summation_method ('scaled_tpm' or 'raw' (default='scaled_tpm'))
fastq_folder (str or Path) – Path to the folder containing the FASTQ files you want to quantify
output_folder (str/Path to an existing folder) – Path to a folder in which the quantified results, as well as the log files, will be saved. The individual output of each pair of FASTQ files will reside in a different sub-folder within the output folder, and a summarized results table will be saved in the output folder itself.
index_file (str or Path) –
Path to a pre-built kallisto index of the target transcriptome. Can either be downloaded from the kallisto transcriptome indices site, or generated manually from a FASTA file using the function kallisto_create_index.
gtf_file (str or Path) – Path to a GTF annotation file. This file will be used to map per-transcript abundances to per-gene estimated counts. The transcript names in the GTF files should match the ones in the index file - we recommend downloading cDNA FASTA/index files and GTF files from the same data source.
average_fragment_length (float > 0) – Estimated average fragment length. Typical Illumina libraries produce fragment lengths ranging from 180–200bp, but it’s best to determine this from a library quantification with an instrument such as an Agilent Bioanalyzer.
stdev_fragment_length (float > 0) – Estimated standard deviation of fragment length. Typical Illumina libraries produce fragment lengths ranging from 180–200bp, but it’s best to determine this from a library quantification with an instrument such as an Agilent Bioanalyzer.
kallisto_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of kallisto. For example: ‘C:/Program Files/kallisto’. if installation folder is set to ‘auto’, RNAlysis will attempt to find it automatically.
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each quantified sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the file pairs.
stranded ('no', 'forward', 'reverse' (default='no')) – Indicates the strandedness of the data. ‘no’ indicates the data is not stranded. ‘forward’ indicates the data is stranded, where the first read in the pair pseudoaligns to the forward strand of a transcript. ‘reverse’ indicates the data is stranded, where the first read in the pair pseudoaligns to the reverse strand of a transcript.
summation_method – Determines the method used to sum the transcript-level abundances to gene-level abundances. ‘scaled_tpm’ sums the transcript TPM estimates the gene level, and then scales then to the library size. ‘raw’ sums the transcript estimated counts to the gene level without scaling.
learn_bias (bool (default=False)) – if True, kallisto learns parameters for a model of sequences specific bias and corrects the abundances accordlingly. Note that this feature is not supported by kallisto versions beyond 0.48.0.
seek_fusion_genes (bool (default=False)) – if True, does normal quantification, but additionally looks for reads that do not pseudoalign because they are potentially from fusion genes. All output is written to the file fusion.txt in the output folder. Note that this feature is not supported by kallisto versions beyond 0.48.0.
bootstrap_samples (int >0 or None (default=None)) – Number of bootstrap samples to be generated. Bootstrap samples do not affect the estimated count values, but generates an additional .hdf5 output file which contains uncertainty estimates for the expression levels. This is required if you use tools such as Sleuth for downstream differential expression analysis, but not for more traditional tools such as DESeq2 and edgeR.
- rnalysis.fastq.sam_to_fastq_paired(input_folder: str | Path, output_folder: str | Path, picard_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', re_reverse_reads: bool = True, include_non_primary_alignments: bool = False, quality_trim: PositiveInt | None = None, return_new_filenames: bool = False)
Convert SAM/BAM files to FASTQ files using Picard SamToFastq.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to convert.
output_folder (str or Path) – Path to a folder in which the converted FASTQ files will be saved.
picard_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of Picard. For example: ‘C:/Program Files/Picard’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each converted sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the files in the directory.
before writing them to FASTQ. :type re_reverse_reads: bool (default=True) :param include_non_primary_alignments: If true, include non-primary alignments in the output. Support of non-primary alignments in SamToFastq is not comprehensive, so there may be exceptions if this is set to true and there are paired reads with non-primary alignments. :type include_non_primary_alignments: bool (default=False) :param quality_trim: If enabled, End-trim reads using the phred/bwa quality trimming algorithm and this quality. :type quality_trim: positive int or None (default=None) :return: a list of the paths to the generated FASTQ files. :return: a list of the paths to the generated FASTQ files. :rtype: list of str
- rnalysis.fastq.sam_to_fastq_single(input_folder: str | Path, output_folder: str | Path, picard_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', re_reverse_reads: bool = True, include_non_primary_alignments: bool = False, quality_trim: PositiveInt | None = None)
Convert SAM/BAM files to FASTQ files using Picard SamToFastq.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to convert.
output_folder (str or Path) – Path to a folder in which the converted FASTQ files will be saved.
picard_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of Picard. For example: ‘C:/Program Files/Picard’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each converted sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the files in the directory.
re_reverse_reads (bool (default=True)) – Re-reverse bases and qualities of reads with the negative-strand flag before writing them to FASTQ.
include_non_primary_alignments (bool (default=False)) – If true, include non-primary alignments in the output. Support of non-primary alignments in SamToFastq is not comprehensive, so there may be exceptions if this is set to true and there are paired reads with non-primary alignments.
quality_trim (positive int or None (default=None)) – If enabled, End-trim reads using the phred/bwa quality trimming algorithm and this quality.
- Returns:
a list of the paths to the generated FASTQ files.
- Return type:
list of str
- rnalysis.fastq.shortstack_align_smallrna(fastq_folder: str | Path, output_folder: str | Path, genome_fasta: str | Path, shortstack_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', known_rnas: str | Path | None = None, trim_adapter: str | Literal['autotrim'] | None = None, autotrim_key: str = 'TCGGACCAGGCTTCATTCCCC', multimap_mode: Literal['fractional', 'unique', 'random'] = 'fractional', align_only: bool = False, show_secondary_alignments: bool = False, dicer_min_length: PositiveInt = 21, dicer_max_length: PositiveInt = 24, loci_file: str | Path | None = None, locus: str | None = None, search_microrna: None | Literal['de-novo', 'known-rnas'] = 'known-rnas', strand_cutoff: Fraction = 0.8, min_coverage: float = 2, pad: PositiveInt = 75, threads: PositiveInt = 1)
Align small RNA single-end reads from FASTQ files to a reference sequence using the ShortStack aligner (version 4). ShortStack is currently not supported on computers running Windows.
- Parameters:
fastq_folder (str or Path) – Path to the folder containing the FASTQ files you want to quantify
output_folder (str/Path to an existing folder) – Path to a folder in which the aligned reads, as well as the log files, will be saved.
genome_fasta (str or Path) – Path to the FASTA file which contain the reference sequences to be aligned to.
shortstack_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of ShortStack. For example: ‘/home/myuser/anaconda3/envs/myenv/bin’. if installation folder is set to ‘auto’, RNAlysis will attempt to find it automatically.
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each quantified sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the file pairs.
known_rnas (str, Path, or None (default=None)) – Path to FASTA-formatted file of known small RNAs. FASTA must be formatted such that a single RNA sequence is on one line only. ATCGUatcgu characters are acceptable. These RNAs are typically the sequences of known microRNAs. For instance, a FASTA file of mature miRNAs pulled from https://www.mirbase.org. Providing these data increases the accuracy of MIRNA locus identification.
trim_adapter (str, 'autotrim', or None (default=None)) – Determines whether ShortStack will attempt to trim the supplied reads. If trim_adapter is not provided (default), no trimming will be run. If trim_adapter is set to ‘autotrim’, ShortStack will automatically infer the 3’ adapter sequence of the untrimmed reads, and the uses that to coordinate read trimming. If trim_adapter is a DNA sequence, ShortStack will trim the reads using the given DNA sequence as the 3’ adapter.
autotrim_key (str (default="TCGGACCAGGCTTCATTCCCC" (miR166))) – A DNA sequence to use as a known suffix during the autotrim procedure. This parameter is used only if trim_adapter is set to ‘autotrim’. ShortStack’s autotrim discovers the 3’ adapter by scanning for reads that begin with the sequence given by autotrim_key. This should be the sequence of a small RNA that is known to be highly abundant in all the libraries. The default sequence is for miR166, a microRNA that is present in nearly all plants at high levels. For non-plant experiments, or if the default is not working well, consider providing an alternative to the default.
multimap_mode ('fractional', 'unique', or 'random' (default='fractional')) – Sets the mode by which multi-mapped reads are handled. These modes are described in Johnson et al. (2016). The default mode (‘fractional’) has the best performance. In ‘fractional’ mode, ShortStack will use a fractional weighting scheme for placement of multi-mapped reads. In ‘unique’ mode, only uniquely-aligned reads are used as weights for placement of multi-mapped reads. In ‘random’ mode, multi-mapped read placement is random.
align_only (bool (default=False)) – if True, ShortStack will terminate after the alignment phase; no additional analysis will occur.
show_secondary_alignments (bool (default=False)) – if True, ShortStack will retain secondary alignments for multimapped reads. This will increase bam file size, possibly by a lot.
dicer_min_length (positive int (default=21)) – the minimum size (in nucleotides) of a valid small RNA. Together with dicer_max_length, this option sets the bounds to discriminate Dicer-derived small RNA loci from other loci. At least 80% of the reads in a given cluster must be in the range indicated by dicer_min_length and dicer_max_length.
dicer_max_length (positive int (default=24)) – the maximun size (in nucleotides) of a valid small RNA. Together with dicer_min_length, this option sets the bounds to discriminate Dicer-derived small RNA loci from other loci. At least 80% of the reads in a given cluster must be in the range indicated by dicer_min_length and dicer_max_length.
loci_file (str, Path, or None (default=None)) – Path to a file of pre-determined loci to analyze. This will prevent de-novo discovery of small RNA loci. The file may be in gff3, bed, or simple tab-delimited format (Chr:Start-Stop[tab]Name). Mutually exclusive with locus.
locus (str or None (default=None)) – A single locus to analyze, given as a string in the format Chr:Start-Stop (using one-based, inclusive numbering). This will prevent de novo discovery of small RNA loci. Mutually exclusive with loci_file.
search_microrna ('de-novo', 'known-rnas', or None (default='known-rnas')) – determines whether and how search for microRNAs will be performed. if search_microrna is None, ShortStack will not search for microRNAs. This saves computational time, but MIRNA loci will not be differentiated from other types of small RNA clusters. if search_microrna is ‘known-rnas’, t ShortStack will confine MIRNA analysis to loci where one or more queries from the known_rnas file are aligned to the genome. if search_microrna is ‘de-novo’, ShortStack will run a more comprehensive genome-wide scan for MIRNA loci. Discovered loci that do not overlap already known microRNAs should be treated with caution.
strand_cutoff (float between 0.5 and 1 (default=0.8)) – Floating point number that sets the cutoff for standedness. By default (strand_cutoff=0.8), loci with >80% reads on the top genomic strand are ‘+’ stranded, loci with <20% reads on the top genomic strand are ‘-’ stranded, and all others are unstranded ‘.’.
min_coverage (float > 0 (default=2)) – Minimum alignment depth, in units of ‘reads per million’, required to nucleate a small RNA cluster during de-novo cluster search.
pad (positive int (default=75)) – initial peaks (continuous regions with depth exceeding the argument min_coverage) are merged if they are this distance or less from each other.
threads (int > 0 (default=1)) – number of threads to run ShortStack on. More threads will generally make index building faster.
- rnalysis.fastq.sort_sam(input_folder: str | Path, output_folder: str | Path, picard_installation_folder: str | Path | Literal['auto'] = 'auto', new_sample_names: List[str] | Literal['auto'] = 'auto', sort_order: Literal['coordinate', 'queryname', 'duplicate'] = 'coordinate')
Sort SAM/BAM files using Picard SortSam.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to sort.
output_folder (str or Path) – Path to a folder in which the sorted SAM/BAM files will be saved.
picard_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of Picard. For example: ‘C:/Program Files/Picard’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each converted sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the files in the directory.
sort_order ('coordinate', 'queryname', or 'duplicate' (default='coordinate')) – The order in which the alignments should be sorted.
- rnalysis.fastq.trim_adapters_paired_end(r1_files: List[str | Path], r2_files: List[str | Path], output_folder: str | Path, three_prime_adapters_r1: None | str | List[str], three_prime_adapters_r2: None | str | List[str], five_prime_adapters_r1: None | str | List[str] = None, five_prime_adapters_r2: None | str | List[str] = None, any_position_adapters_r1: None | str | List[str] = None, any_position_adapters_r2: None | str | List[str] = None, new_sample_names: List[str] | Literal['auto'] = 'auto', quality_trimming: NonNegativeInt | None = 20, trim_n: bool = True, minimum_read_length: NonNegativeInt = 10, maximum_read_length: PositiveInt | None = None, discard_untrimmed_reads: bool = True, pair_filter_if: Literal['both', 'any', 'first'] = 'both', error_tolerance: Fraction = 0.1, minimum_overlap: NonNegativeInt = 3, allow_indels: bool = True, parallel: bool = True, gzip_output: bool = False, return_new_filenames: bool = False)
Trim adapters from paired-end reads using CutAdapt.
- Parameters:
r1_files (list of str/Path to existing FASTQ files) – a list of paths to your Read#1 files. The files should be sorted in tandem with r2_files, so that they line up to form pairs of R1 and R2 files.
r2_files (list of str/Path to existing FASTQ files) – a list of paths to your Read#2 files. The files should be sorted in tandem with r1_files, so that they line up to form pairs of R1 and R2 files.
output_folder (str/Path to an existing folder) – Path to a folder in which the trimmed FASTQ files, as well as the log files, will be saved.
three_prime_adapters_r1 (str, list of str, or None) – the sequence of the adapter/adapters to trim from the 3’ end of the reads in Read#1 files.
three_prime_adapters_r2 (str, list of str, or None) – the sequence of the adapter/adapters to trim from the 3’ end of the reads in Read#2 files.
five_prime_adapters_r1 (str, list of str, or None (default=None)) – the sequence of the adapter/adapters to trim from the 5’ end of the reads in Read#1 files.
five_prime_adapters_r2 (str, list of str, or None (default=None)) – the sequence of the adapter/adapters to trim from the 5’ end of the reads in Read#2 files.
any_position_adapters_r1 (str, list of str, or None (default=None)) – the sequence of the adapter/adapters to trim from either end (or the middle) of the reads in Read#1 files.
any_position_adapters_r2 (str, list of str, or None (default=None)) – the sequence of the adapter/adapters to trim from either end (or the middle) of the reads in Read#2 files.
quality_trimming (int or None (default=20)) – if specified, trim low-quality 3’ end from the reads. Any bases with quality score below the specified value will be trimmed from the 3’ end of the read.
trim_n (bool (default=True)) – if True, removem flanking N bases from each read. For example, a read with the sequence ‘NNACGTACGTNNNN’ will be trimmed down to ‘ACGTACGT’. This occurs after adapter trimming.
minimum_read_length (int or None (default=10)) – if specified (default), discard processed reads that are shorter than minimum_read_length.
maximum_read_length (int or None (default=None)) – if specified, discard processed reads that are shorter than minimum_read_length.
discard_untrimmed_reads (bool (default=True)) – if True, discards reads in which no adapter was found.
pair_filter_if ('both', 'any', or 'first' (default='both')) – Cutadapt always discards both reads of a pair if it determines that the pair should be discarded. This parameter determines how to combine the filters for Read#1 and Read#2 into a single decision about the read pair. When the value is ‘both’, you require that filtering criteria must apply to both reads in order for a read pair to be discarded. When the value is ‘any’, you require that at least one of the reads (R1 or R2) fulfills the filtering criterion in order to discard them. When the value is ‘first’, only the first read in each pair determines whether to discard the pair or not.
error_tolerance (float between 0 and 1 (default=0.1)) – The level of error tolerance permitted when searching for adapters, with the lowest value being 0 (no error tolerance) and the maximum being 1 (100% error tolerance). Allowed errors are mismatches, insertions and deletions.
minimum_overlap (int >= 0 (default=3)) – the minimum number of nucleotides that must match exactly to the adapter sequence in order to trim it.
allow_indels (bool (default=True)) – if False, insertions and deletions in the adapter sequence are not allowed - only mismatches.
parallel (bool (default=True)) – if True, runs CutAdapt on all available cores in parallel. Otherwise, run CutAdapt on a single processor only.
gzip_output (bool (default=False)) – if True, gzips the output FASTQ files.
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each trimmed sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the file pairs.
- rnalysis.fastq.trim_adapters_single_end(fastq_folder: str | Path, output_folder: str | Path, three_prime_adapters: None | str | List[str], five_prime_adapters: None | str | List[str] = None, any_position_adapters: None | str | List[str] = None, new_sample_names: List[str] | Literal['auto'] = 'auto', quality_trimming: NonNegativeInt | None = 20, trim_n: bool = True, minimum_read_length: NonNegativeInt = 10, maximum_read_length: PositiveInt | None = None, discard_untrimmed_reads: bool = True, error_tolerance: Fraction = 0.1, minimum_overlap: NonNegativeInt = 3, allow_indels: bool = True, parallel: bool = True, gzip_output: bool = False)
Trim adapters from single-end reads using CutAdapt.
- Parameters:
fastq_folder (str/Path to an existing folder) – Path to the folder containing your untrimmed FASTQ files
output_folder (str/Path to an existing folder) – Path to a folder in which the trimmed FASTQ files, as well as the log files, will be saved.
three_prime_adapters (str, list of str, or None) – the sequence of the adapter/adapters to trim from the 3’ end of the reads.
five_prime_adapters (str, list of str, or None (default=None)) – the sequence of the adapter/adapters to trim from the 5’ end of the reads.
any_position_adapters (str, list of str, or None (default=None)) – the sequence of the adapter/adapters to trim from either end (or from the middle) of the reads.
quality_trimming (int or None (default=20)) – if specified, trim low-quality 3’ end from the reads. Any bases with quality score below the specified value will be trimmed from the 3’ end of the read.
trim_n (bool (default=True)) – if True, removem flanking N bases from each read. For example, a read with the sequence ‘NNACGTACGTNNNN’ will be trimmed down to ‘ACGTACGT’. This occurs after adapter trimming.
minimum_read_length (int or None (default=10)) – if specified (default), discard processed reads that are shorter than minimum_read_length.
maximum_read_length (int or None (default=None)) – if specified, discard processed reads that are shorter than minimum_read_length.
discard_untrimmed_reads (bool (default=True)) – if True, discards reads in which no adapter was found.
error_tolerance (float between 0 and 1 (default=0.1)) – The level of error tolerance permitted when searching for adapters, with the lowest value being 0 (no error tolerance) and the maximum being 1 (100% error tolerance). Allowed errors are mismatches, insertions and deletions.
minimum_overlap (int >= 0 (default=3)) – the minimum number of nucleotides that must match exactly to the adapter sequence in order to trim it.
allow_indels (bool (default=True)) – if False, insertions and deletions in the adapter sequence are not allowed - only mismatches.
parallel (bool (default=True)) – if True, runs CutAdapt on all available cores in parallel. Otherwise, run CutAdapt on a single processor only.
gzip_output (bool (default=False)) – if True, gzips the output FASTQ files.
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each trimmed sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the alphabetical order of the input files.
- rnalysis.fastq.validate_sam(input_folder: str | Path, output_folder: str | Path, picard_installation_folder: str | Path | Literal['auto'] = 'auto', verbose: bool = True, is_bisulfite_sequenced: bool = False)
Validate SAM/BAM files using Picard ValidateSamFile.
- Parameters:
input_folder (str or Path) – Path to the folder containing the SAM/BAM files you want to validate.
output_folder (str or Path) – Path to a folder in which the validation reports will be saved.
picard_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of Picard. For example: ‘C:/Program Files/Picard’
new_sample_names (list of str or 'auto' (default='auto')) – Give a new name to each converted sample (optional). If sample_names=’auto’, sample names will be given automatically. Otherwise, sample_names should be a list of new names, with the order of the names matching the order of the files in the directory.
verbose (bool (default=True)) – If True, the validation report will be verbose. If False, the validation report will be a summary.
is_bisulfite_sequenced (bool (default=False)) – Indicates whether the SAM/BAM file consists of bisulfite sequenced reads. If so, C->T is not counted as en error in computer the value of the NM tag.
- Returns:
a list of the paths to the generated FASTQ files.
- Return type:
list of str
rnalysis.filtering module
This module can filter, normalize, intersect and visualize tabular data such as read counts and differential expression data. Any tabular data saved in a csv format can be imported. Use this module to perform various filtering operations on your data, normalize your data, perform set operations (union, intersection, etc), run basic exploratory analyses and plots (such as PCA, clustergram, violin plots, scatter, etc), save the filtered data to your computer, and more. When you save filtered/modified data, its new file name will include by default all of the operations performed on it, in the order they were performed, to allow easy traceback of your analyses.
- class rnalysis.filtering.CountFilter(fname: str | Path | tuple, drop_columns: str | List[str] = None, is_normalized: bool = False, suppress_warnings: bool = False)
Bases:
FilterA class that receives a count matrix and can filter it according to various characteristics.
Attributes
- df: polars DataFrame
A DataFrame that contains the count matrix contents. The DataFrame is modified upon usage of filter operations.
- shape: tuple (rows, columns)
The dimensions of df.
- columns: list
The columns of df.
- fname: pathlib.Path
The path and filename for the purpose of saving df as a csv file. Updates automatically when filter operations are applied.
- index_set: set
All of the indices in the current DataFrame (which were not removed by previously used filter methods) as a set.
- index_string: string
A string of all feature indices in the current DataFrame separated by newline.
- triplicates: list
Returns a nested list of the column names in the CountFilter, grouped by alphabetical order into triplicates. For example, if counts.columns is [‘A_rep1’,’A_rep2’,’A_rep3’,’B_rep1’,’B_rep2’,_B_rep3’], then counts.triplicates will be [[‘A_rep1’,’A_rep2’,’A_rep3’],[‘B_rep1’,’B_rep2’,_B_rep3’]]
- _avg_subsamples(sample_grouping: GroupedColumns, function: Literal['mean', 'median', 'geometric_mean'] = 'mean', new_column_names: Literal['auto'] | Literal['display'] | List[str] = 'display')
Avarages subsamples/replicates according to the specified sample list. Every member in the sample list should be either a name of a single sample (str), or a list of multiple sample names to be averaged (list).
- Parameters:
sample_grouping – A list of the sample names and/or grouped sample names passed by the user. All specified samples must be present in the CountFilter object. To average multiple replicates of the same condition, they can be grouped in an inner list. Example input: [[‘SAMPLE1A’, ‘SAMPLE1B’, ‘SAMPLE1C’], [‘SAMPLE2A’, ‘SAMPLE2B’, ‘SAMPLE2C’],’SAMPLE3’ , ‘SAMPLE6’] and the resulting output will be a DataFrame containing the following columns: [‘SAMPLE1’, ‘SAMPLE2’, ‘SAMPLE3’, ‘SAMPLE6’]
- Returns:
a polars DataFrame containing samples/averaged subsamples according to the specified sample_list.
- static _from_string(msg: str = '', delimiter: str = '\n')
Takes a manual string input from the user, and then splits it using a delimiter into a list of values.
- param msg:
a promprt to be printed to the user
- param delimiter:
the delimiter used to separate the values. Default is ‘
- ‘
- return:
A list of the comma-seperated values the user inserted.
- _inplace(new_df: DataFrame, opposite: bool, inplace: bool, suffix: str, printout_operation: str = 'filter', **filter_update_kwargs)
Executes the user’s choice whether to filter in-place or create a new instance of the Filter object.
- Parameters:
new_df (pl.DataFrame) – the post-filtering DataFrame
opposite (bool) – Determines whether to return the filtration ,or its opposite.
inplace (bool) – Determines whether to filter in-place or not.
suffix (str) – The suffix to be added to the filename
- Returns:
If inplace is False, returns a new instance of the Filter object.
- _is_normalized
indicates whether the values in this CountFilter were normalized
- property _numeric_columns: list
Returns a list of the numeric (int/float) columns in the DataFrame.
- static _pca_plot(final_df: DataFrame, pc1_var: float, pc2_var: float, sample_grouping: GroupedColumns, labels: bool, title: str, title_fontsize: float, label_fontsize: float, tick_fontsize: float, proportional_axes: bool, plot_grid: bool, legend: List[str] | None) Figure
Internal method, used to plot the results from CountFilter.pca().
- Parameters:
final_df – The DataFrame output from pca
pc1_var – Variance explained by the first PC.
pc2_var – Variance explained by the second PC.
sample_grouping – A list of the sample names and/or grouped sample names to be plotted. All specified samples must be present in the DataFrame object. To draw multiple replicates of the same condition in the same color, they can be grouped in an inner list. Example input: [[‘SAMPLE1A’, ‘SAMPLE1B’, ‘SAMPLE1C’], [‘SAMPLE2A’, ‘SAMPLE2B’, ‘SAMPLE2C’],’SAMPLE3’ , ‘SAMPLE6’]
- Returns:
an axis object containing the PCA plot.
- _set_ops(others, return_type: Literal['set', 'str'], op: Any, **kwargs)
Apply the supplied set operation (union/intersection/difference/symmetric difference) to the supplied objects.
- Parameters:
others (Filter or set objects.) – the other objects to apply the set operation to
return_type ('set' or 'str') – the return type of the output
op (function (set.union, set.intersection, set.difference or set.symmetric_difference)) – the set operation
kwargs – any additional keyworded arguments to be supplied to the set operation.
- Returns:
a set/string of indices resulting from the set operation
- Return type:
set or str
- _sort(by: str | List[str], ascending: bool | List[bool] = True, na_position: str = 'last')
Sort the rows by the values of specified column or columns.
- Parameters:
by (str or list of str) – Names of the column or columns to sort by.
ascending (bool or list of bool, default True) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
inplace (bool, default True) – If True, perform operation in-place. Otherwise, returns a sorted copy of the Filter object without modifying the original.
- Returns:
None if inplace=True, a sorted Filter object otherwise.
- annotate_from_gtf(gtf_path: str | Path, attribute: Literal['gene_biotype', 'transcript_biotype', 'biotype', 'gene_name', 'gene_id', 'transcript_id', 'chromosome', 'source', 'strand'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', column_name: str | None = None, inplace: bool = True)
Adds a new column to the table, annotating each feature with the value of a GTF/GFF attribute (for example: the biotype, chromosome, strand, or source of each gene). Features that are not found in the annotation file are annotated with a missing value.
- Parameters:
gtf_path (str or Path) – Path to your GTF/GFF annotation file. The file should match the type of gene names/IDs you use in your table.
attribute (str (default='gene_biotype')) – name of the attribute to annotate with. Standard column-9 attributes (such as ‘gene_biotype’, ‘gene_name’, or any custom key in your file) are supported, as well as the reserved names ‘chromosome’, ‘source’ and ‘strand’, which are read from the fixed columns of the annotation file.
feature_type ('gene' or 'transcript' (default='gene')) – determines whether the features/rows in your data table describe individual genes or transcripts.
column_name (str or None (default=None)) – the name of the new annotation column. If not specified, the attribute name is used. If a column with this name already exists in the table, it will be overwritten.
inplace (bool (default=True)) – If True (default), the annotation column will be added to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and annotated instance of the Filter object.
- average_replicate_samples(sample_grouping: GroupedColumns, new_column_names: Literal['auto'] | List[str] = 'auto', function: Literal['mean', 'median', 'geometric_mean'] = 'mean', inplace: bool = True) CountFilter
Average the expression values of gene expression for each group of replicate samples. Each group of samples (e.g. biological/technical replicates)
- Parameters:
sample_grouping (nested list of column names) – grouping of the samples into conditions. Each grouping should containg all replicates of the same condition. Each condition will be averaged separately.
new_column_names (list of str or 'auto' (default='auto') – names to be given to the columns in the new count matrix. Each new name should match a group of samples to be averaged. If `new_column_names`=’auto’, names will be generated automatically.
function ('mean', 'median', or 'geometric_mean' (default='mean')) – the function which will be used to average the values within each group.
inplace (bool (default=True)) – If True (default), averaging will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
- biotypes_from_gtf(gtf_path: str | Path, attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', long_format: bool = False) DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- biotypes_from_ref_table(long_format: bool = False, ref: str | Path | Literal['predefined'] = 'predefined') DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :param ref: Name of the biotype reference table used to determine biotype. Default is ce11 (included in the package). :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # short-form view >>> d.biotypes_from_ref_table(ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 2) ┌────────────────┬───────┐ │ biotype ┆ count │ │ --- ┆ --- │ │ str ┆ u32 │ ╞════════════════╪═══════╡ │ unknown ┆ 1 │ │ protein_coding ┆ 26 │ │ pseudogene ┆ 1 │ └────────────────┴───────┘
>>> # long-form view >>> d.biotypes_from_ref_table(long_format=True,ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 49) ┌───────────┬───────────┬───────────┬───────────┬───┬───────────┬───────────┬───────────┬──────────┐ │ biotype ┆ baseMean_ ┆ baseMean_ ┆ baseMean_ ┆ … ┆ padj_25% ┆ padj_50% ┆ padj_75% ┆ padj_max │ │ --- ┆ count ┆ mean ┆ std ┆ ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ --- ┆ --- ┆ --- ┆ ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ │ ┆ f64 ┆ f64 ┆ f64 ┆ ┆ ┆ ┆ ┆ │ ╞═══════════╪═══════════╪═══════════╪═══════════╪═══╪═══════════╪═══════════╪═══════════╪══════════╡ │ protein_c ┆ 26.0 ┆ 1823.0896 ┆ 1796.5207 ┆ … ┆ 3.8506e-1 ┆ 1.1350e-1 ┆ 1.0051e-9 ┆ 9.2900e- │ │ oding ┆ ┆ 09 ┆ 8 ┆ ┆ 76 ┆ 35 ┆ 0 ┆ 68 │ │ pseudogen ┆ 1.0 ┆ 2688.0437 ┆ null ┆ … ┆ 1.8000e-9 ┆ 1.8000e-9 ┆ 1.8000e-9 ┆ 1.8000e- │ │ e ┆ ┆ 01 ┆ ┆ ┆ 4 ┆ 4 ┆ 4 ┆ 94 │ │ unknown ┆ 1.0 ┆ 2085.9950 ┆ null ┆ … ┆ 3.0700e-1 ┆ 3.0700e-1 ┆ 3.0700e-1 ┆ 3.0700e- │ │ ┆ ┆ 94 ┆ ┆ ┆ 52 ┆ 52 ┆ 52 ┆ 152 │ └───────────┴───────────┴───────────┴───────────┴───┴───────────┴───────────┴───────────┴──────────┘
- box_plot(samples: GroupedColumns | Literal['all'] = 'all', notch: bool = True, scatter: bool = False, ylabel: str = 'log10(Normalized reads + 1)', title: str | Literal['auto'] = 'auto', title_fontsize: float = 20, label_fontsize: float = 16, tick_fontsize: float = 12) Figure
Generates a box plot of the specified samples in the CountFilter object in log10 scale. Can plot both single samples and average multiple replicates. It is recommended to use this function on normalized values and not on absolute read values. The box indicates 25% and 75% percentiles, and the white dot indicates the median.
- Parameters:
samples ('all' or list.) – A list of the sample names and/or grouped sample names to be plotted. All specified samples must be present in the CountFilter object. To average multiple replicates of the same condition, they can be grouped in an inner list. Example input: [[‘SAMPLE1A’, ‘SAMPLE1B’, ‘SAMPLE1C’], [‘SAMPLE2A’, ‘SAMPLE2B’, ‘SAMPLE2C’],’SAMPLE3’ , ‘SAMPLE6’]
notch (bool (default=True)) – if True, adds a confidence-interval notch to the box-plot.
scatter (bool (default=False)) – if True, adds a scatter-plot on top of the box-plot.
ylabel (str (default='Log10(Normalized reads + 1)')) – the label of the Y axis.
title (str or 'auto' (default='auto')) – The title of the plot. If ‘auto’, a title will be generated automatically.
title_fontsize (float (default=30)) – determines the font size of the graph title.
label_fontsize (float (default=15) :param tick_fontsize: determines the font size of the X and Y tick labels.) – determines the font size of the X and Y axis labels.
- Return type:
a matplotlib Figure.
Example plot of box_plot(add_scatter=True)
- clustergram(sample_names: ColumnNames | Literal['all'] = 'all', metric: Literal['Correlation', 'Cosine', 'Euclidean', 'Jaccard'] | str = 'Euclidean', linkage: Literal['Single', 'Average', 'Complete', 'Ward', 'Weighted', 'Centroid', 'Median'] = 'Average', title: str | Literal['auto'] = 'auto', title_fontsize: float = 20, tick_fontsize: float = 12, colormap: ColorMap = 'inferno', colormap_label: Literal['auto'] | str = 'auto', cluster_columns: bool = True, log_transform: bool = True, z_score_rows: bool = False) Figure
Performs hierarchical clustering and plots a clustergram on the base-2 log of a given set of samples.
- Parameters:
z_score_rows (bool (default=False)) – if True, the rows will be z-scored before clustering. This will normalize the rows to have a mean of 0 and a standard deviation of 1, such that genes will be clustered based on the similarity of their expression pattern instead of absolute expression levels.
colormap_label (str or 'auto' (default='auto')) – label for the colorbar
cluster_columns (bool (default=True)) – if True, both rows and columns will be clustered. Otherwise, only the rows will be clustered, and columns will maintain their original order.
colormap (str) – the colormap to use in the clustergram.
log_transform (bool (default=True)) – if True, will apply a log transform (log2) to the data before clustering.
sample_names ('all' or list.) – the names of the relevant samples in a list. Example input: [“condition1_rep1”, “condition1_rep2”, “condition1_rep3”, “condition2_rep1”, “condition3_rep1”, “condition3_rep2”]
metric ('Euclidean', 'hamming', 'correlation', or any other distance metric available in scipy.spatial.distance.pdist) – the distance metric to use in the clustergram. For all possible inputs and their meaning see scipy.spatial.distance.pdist documentation online.
linkage ('single', 'average', 'complete', 'weighted', 'centroid', 'median' or 'ward'.) – the linkage method to use in the clustergram. For all possible inputs and their meaning see scipy.cluster.hierarchy.linkage documentation online.
title (str or 'auto' (default='auto')) – The title of the plot. If ‘auto’, a title will be generated automatically.
title_fontsize (float (default=30)) – determines the font size of the graph title.
tick_fontsize (float (default=10)) – determines the font size of the X and Y tick labels.
- Return type:
A matplotlib Figure.
Example plot of clustergram()
- property columns: list
The columns of df.
- Returns:
a list of the columns in the Filter object.
- Return type:
list
- describe(percentiles: float | List[float] = (0.01, 0.25, 0.5, 0.75, 0.99)) DataFrame
Generate descriptive statistics that summarize the central tendency, dispersion and shape of the dataset’s distribution, excluding NaN values. For more information see the documentation of polars.DataFrame.describe.
- Parameters:
percentiles (list-like of floats (default=(0.01, 0.25, 0.5, 0.75, 0.99))) – The percentiles to include in the output. All should fall between 0 and 1. The default is [.25, .5, .75], which returns the 25th, 50th, and 75th percentiles.
- Returns:
Summary statistics of the dataset.
- Return type:
pl.DataFrame
- Examples:
>>> from rnalysis import filtering >>> import numpy as np >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.describe() cond1 cond2 cond3 cond4 count 22.000000 22.000000 22.000000 22.000000 mean 2515.590909 2209.227273 4230.227273 3099.818182 std 4820.512674 4134.948493 7635.832664 5520.394522 min 0.000000 0.000000 0.000000 0.000000 1% 0.000000 0.000000 0.000000 0.000000 25% 6.000000 6.250000 1.250000 0.250000 50% 57.500000 52.500000 23.500000 21.000000 75% 2637.000000 2479.000000 6030.500000 4669.750000 99% 15054.950000 12714.290000 21955.390000 15603.510000 max 15056.000000 12746.000000 22027.000000 15639.000000
>>> # show the deciles (10%, 20%, 30%... 90%) of the columns >>> counts.describe(percentiles=np.arange(0.1, 1, 0.1)) cond1 cond2 cond3 cond4 count 22.000000 22.000000 22.000000 22.000000 mean 2515.590909 2209.227273 4230.227273 3099.818182 std 4820.512674 4134.948493 7635.832664 5520.394522 min 0.000000 0.000000 0.000000 0.000000 10% 0.000000 0.200000 0.000000 0.000000 20% 1.400000 3.200000 1.000000 0.000000 30% 15.000000 15.700000 2.600000 1.000000 40% 28.400000 26.800000 14.000000 9.000000 50% 57.500000 52.500000 23.500000 21.000000 60% 82.000000 106.800000 44.000000 33.000000 70% 484.200000 395.500000 305.000000 302.500000 80% 3398.600000 3172.600000 7981.400000 6213.000000 90% 8722.100000 7941.800000 16449.500000 12129.900000 max 15056.000000 12746.000000 22027.000000 15639.000000
- df
- difference(*others: Filter | set, return_type: Literal['set', 'str'] = 'set', inplace: bool = False)
Keep only the features that exist in the first Filter object/set but NOT in the others. Can be done inplace on the first Filter object, or return a set/string of features.
- Parameters:
others (Filter or set objects.) – Objects to calculate difference with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the features that exist only in the first Filter object. If ‘str’, returns a string of the WBGene indices that exist only in the first Filter object, delimited by a comma.
inplace (bool, default False) – If True, filtering will be applied to the current Filter object. If False (default), the function will return a set/str that contains the intersecting features.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that exist only in the first Filter object/set (set difference).
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter("tests/test_files/test_deseq.csv") >>> counts = filtering.CountFilter('tests/test_files/counted.csv') >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> # calculate difference and return a set >>> d.difference(counts, a_set) {'WBGene00007063', 'WBGene00007064', 'WBGene00007066', 'WBGene00007067', 'WBGene00007069', 'WBGene00007071', 'WBGene00007074', 'WBGene00007075', 'WBGene00007076', 'WBGene00007077', 'WBGene00007078', 'WBGene00007079', 'WBGene00014997', 'WBGene00043987', 'WBGene00043988', 'WBGene00043989', 'WBGene00043990', 'WBGene00044022', 'WBGene00044951', 'WBGene00077502', 'WBGene00077503', 'WBGene00077504'}
# calculate difference and filter in-place >>> d.difference(counts, a_set, inplace=True) Filtered 2 features, leaving 26 of the original 28 features. Filtered inplace.
- differential_expression_deseq2(design_matrix: str | Path, comparisons: Iterable[Tuple[str, str, str]], covariates: Iterable[str] = (), lrt_factors: Iterable[str] = (), model_factors: Literal['auto'] | Iterable[str] = 'auto', r_installation_folder: str | Path | Literal['auto'] = 'auto', output_folder: str | Path | None = None, return_design_matrix: bool = False, scaling_factors: str | Path | None = None, cooks_cutoff: bool = True, return_code: bool = False, return_log: bool = False) Tuple[DESeqFilter, ...]
Run differential expression analysis on the count matrix using the DESeq2 algorithm. The count matrix you are analyzing should be unnormalized (meaning, raw read counts). The analysis will be based on a design matrix supplied by the user. The design matrix should contain at least two columns: the first column contains all the sample names, and each of the following columns contains an experimental design factor (e.g. ‘condition’, ‘replicate’, etc). (see the User Guide and Tutorial for a complete example). The analysis formula will contain all the factors in the design matrix. To run this function, a version of R must be installed.
- Parameters:
design_matrix (str or Path) – path to a csv file containing the experiment’s design matrix. The design matrix should contain at least two columns: the first column contains all the sample names, and each of the following columns contains an experimental design factor (e.g. ‘condition’, ‘replicate’, etc). (see the User Guide and Tutorial for a complete example). The analysis formula will contain all the factors in the design matrix.
comparisons (Iterable of tuple(factor, numerator_value, denominator_value)) – specifies what comparisons to build results tables out of. each individual comparison should be a tuple with exactly three elements: the name of a factor in the design formula, the name of the numerator level for the fold change, and the name of the denominator level for the fold change.
lrt_factors (Iterable of factor names (default=tuple())) – optionally, specify factors to be tested using the likelihood ratio test (LRT). If the factors are a continuous variable, you can also specify the number of polynomial degree to fit.
covariates (Iterable of covariate names (default=tuple())) – optionally, specify a list of continuous covariates to include in the analysis. The covariates should be column names in the design matrix. The reported fold change values correspond to the expected fold change for every increase of 1 unit in the covariate.
model_factors (Iterable of factor names or 'auto' (default='auto')) – optionally, specify a list of factors to include in the differential expression model. If ‘auto’, all factors in the design matrix will be included.
r_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of R. For example: ‘C:/Program Files/R/R-4.2.1’
output_folder (str, Path, or None) – Path to a folder in which the analysis results, as well as the log files and R script used to generate them, will be saved. if output_folder is None, the results will not be saved to a specified directory.
return_design_matrix (bool (default=False)) – if True, the function will return the sanitized design matrix used in the analysis.
return_code (bool (default=False)) – if True, the function will return the path to the R script used to generate the analysis results.
return_log (bool (default=False)) – if True, the function will return the path to the analysis logfile, which includes session info.
- Returns:
a tuple of DESeqFilter objects, one for each comparison
- differential_expression_deseq2_simplified(design_matrix: str | Path, comparisons: Iterable[Tuple[str, str, str]], r_installation_folder: str | Path | Literal['auto'] = 'auto', output_folder: str | Path | None = None, return_design_matrix: bool = False, return_code: bool = False, return_log: bool = False) Tuple[DESeqFilter, ...]
Run differential expression analysis on the count matrix using the DESeq2 algorithm. The simplified mode supports only pairwise comparisons. The count matrix you are analyzing should be unnormalized (meaning, raw read counts). The analysis will be based on a design matrix supplied by the user. The design matrix should contain at least two columns: the first column contains all the sample names, and each of the following columns contains an experimental design factor (e.g. ‘condition’, ‘replicate’, etc). (see the User Guide and Tutorial for a complete example). The analysis formula will contain all the factors in the design matrix. To run this function, a version of R must be installed.
- Parameters:
design_matrix (str or Path) – path to a csv file containing the experiment’s design matrix. The design matrix should contain at least two columns: the first column contains all the sample names, and each of the following columns contains an experimental design factor (e.g. ‘condition’, ‘replicate’, etc). (see the User Guide and Tutorial for a complete example). The analysis formula will contain all the factors in the design matrix.
comparisons (Iterable of tuple(factor, numerator_value, denominator_value)) – specifies what comparisons to build results tables out of. each individual comparison should be a tuple with exactly three elements: the name of a factor in the design formula, the name of the numerator level for the fold change, and the name of the denominator level for the fold change.
r_installation_folder (str, Path, or 'auto' (default='auto')) – Path to the installation folder of R. For example: ‘C:/Program Files/R/R-4.2.1’
output_folder (str, Path, or None) – Path to a folder in which the analysis results, as well as the log files and R script used to generate them, will be saved. if output_folder is None, the results will not be saved to a specified directory.
return_design_matrix (bool (default=False)) – if True, the function will return the sanitized design matrix used in the analysis.
return_code (bool (default=False)) – if True, the function will return the R script used to generate the analysis results.
- Returns:
a tuple of DESeqFilter objects, one for each comparison
- differential_expression_limma_voom(design_matrix: str | Path, comparisons: Iterable[Tuple[str, str, str]], covariates: Iterable[str] = (), lrt_factors: Iterable[str] = (), model_factors: Literal['auto'] | Iterable[str] = 'auto', r_installation_folder: str | Path | Literal['auto'] = 'auto', output_folder: str | Path | None = None, random_effect: str | None = None, quality_weights: bool = False, return_design_matrix: bool = False, return_code: bool = False, return_log: bool = False) Tuple[DESeqFilter, ...]
Run differential expression analysis on the count matrix using the Limma-Voom pipeline. The count matrix you are analyzing should be normalized (typically to Reads Per Million). The analysis will be based on a design matrix supplied by the user. The design matrix should contain at least two columns: the first column contains all the sample names, and each of the following columns contains an experimental design factor (e.g. ‘condition’, ‘replicate’, etc). (see the User Guide and Tutorial for a complete example). The analysis formula will contain all the factors in the design matrix. To run this function, a version of R must be installed.
- param design_matrix:
path to a csv file containing the experiment’s design matrix. The design matrix should contain at least two columns: the first column contains all the sample names, and each of the following columns contains an experimental design factor (e.g. ‘condition’, ‘replicate’, etc). (see the User Guide and Tutorial for a complete example). The analysis formula will contain all the factors in the design matrix.
- type design_matrix:
str or Path
- Parameters:
comparisons – specifies what comparisons to build results tables out of. each individual comparison should be a tuple with exactly three elements: the name of a factor in the design formula, the name of the numerator level for the fold change, and the name of the denominator level for the fold change. :type comparisons: Iterable of tuple(factor, numerator_value, denominator_value) :param lrt_factors: optionally, specify factors to be tested using the likelihood ratio test (LRT). If the factors are a continuous variable, you can also specify the number of polynomial degree to fit. :type lrt_factors: Iterable of tuple(factor, polynomial_degree) or None (default=None) :param covariates: optionally, specify a list of continuous covariates to include in the analysis. The covariates should be column names in the design matrix. The reported fold change values correspond to the expected fold change for every increase of 1 unit in the covariate. :param model_factors: optionally, specify a list of factors to include in the differential expression model. If ‘auto’, all factors in the design matrix will be included. :type model_factors: Iterable of factor names or ‘auto’ (default=’auto’) :param r_installation_folder: Path to the installation folder of R. For example: ‘C:/Program Files/R/R-4.2.1’ :type r_installation_folder: str, Path, or ‘auto’ (default=’auto’) :param output_folder: Path to a folder in which the analysis results, as well as the log files and R script used to generate them, will be saved. if output_folder is None, the results will not be saved to a specified directory. :type output_folder: str, Path, or None :param random_effect: optionally, specify a single factor to model as a random effect. This is useful when your experimental design is nested. limma-voom can only treat one factor as a random effect. :type random_effect: str or None :param quality_weights: if True, the analysis will use estimate sample-specific quality weights using the ‘arrayWeights’ function im limma. This is useful when lower quality samples are present in the data. :type quality_weights: bool (default=False) :param return_design_matrix: if True, the function will return the sanitized design matrix used in the analysis. :type return_design_matrix: bool (default=False) :param return_code: if True, the function will return the path to the R script used to generate the analysis results. :type return_code: bool (default=False) :param return_log: if True, the function will return the path to the analysis logfile, which includes session info. :type return_log: bool (default=False) :return: a tuple of DESeqFilter objects, one for each comparison
- differential_expression_limma_voom_simplified(design_matrix: str | Path, comparisons: Iterable[Tuple[str, str, str]], r_installation_folder: str | Path | Literal['auto'] = 'auto', output_folder: str | Path | None = None, random_effect: str | None = None, quality_weights: bool = False, return_design_matrix: bool = False, return_code: bool = False, return_log: bool = False) Tuple[DESeqFilter, ...]
Run differential expression analysis on the count matrix using the Limma-Voom pipeline. The simplified mode supports only pairwise comparisons. The count matrix you are analyzing should be normalized (typically to Reads Per Million). The analysis will be based on a design matrix supplied by the user. The design matrix should contain at least two columns: the first column contains all the sample names, and each of the following columns contains an experimental design factor (e.g. ‘condition’, ‘replicate’, etc). (see the User Guide and Tutorial for a complete example). The analysis formula will contain all the factors in the design matrix. To run this function, a version of R must be installed.
- param design_matrix:
path to a csv file containing the experiment’s design matrix. The design matrix should contain at least two columns: the first column contains all the sample names, and each of the following columns contains an experimental design factor (e.g. ‘condition’, ‘replicate’, etc). (see the User Guide and Tutorial for a complete example). The analysis formula will contain all the factors in the design matrix.
- type design_matrix:
str or Path
- Parameters:
comparisons –
specifies what comparisons to build results tables out of. each individual comparison should be a tuple with exactly three elements: the name of a factor in the design formula, the name of the numerator level for the fold change, and the name of the denominator level for the fold change. :type comparisons: Iterable of tuple(factor, numerator_value, denominator_value) :param r_installation_folder: Path to the installation folder of R. For example: ‘C:/Program Files/R/R-4.2.1’ :type r_installation_folder: str, Path, or ‘auto’ (default=’auto’) :param output_folder: Path to a folder in which the analysis results, as well as the log files and R script used to generate them, will be saved. if output_folder is None, the results will not be saved to a specified directory. :type output_folder: str, Path, or None :param random_effect: optionally, specify a single factor to model as a random effect. This is useful when your experimental design is nested. limma-voom can only treat one factor as a random effect. :type random_effect: str or None :param quality_weights: if True, the analysis will use estimate sample-specific quality weights using the ‘arrayWeights’ function im limma. This is useful when lower quality samples are present in the data.
- type quality_weights:
bool (default=False)
- param return_design_matrix:
if True, the function will return the sanitized design matrix used in the analysis.
- type return_design_matrix:
bool (default=False)
- param return_code:
if True, the function will return the R script used to generate the analysis results.
- type return_code:
bool (default=False)
- return:
a tuple of DESeqFilter objects, one for each comparison
- drop_columns(columns: ColumnNames, inplace: bool = True)
Drop specific columns from the table.
- Parameters:
columns (str or list of str) – The names of the column/columns to be dropped fro mthe table.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- enhanced_box_plot(samples: GroupedColumns | Literal['all'] = 'all', scatter: bool = False, ylabel: str = 'log10(Normalized reads + 1)', title: str | Literal['auto'] = 'auto', title_fontsize: float = 20, label_fontsize: float = 16, tick_fontsize: float = 12) Figure
Generates an enhanced box-plot of the specified samples in the CountFilter object in log10 scale. Can plot both single samples and average multiple replicates. It is recommended to use this function on normalized values and not on absolute read values. The box indicates 25% and 75% percentiles, and the white dot indicates the median.
- Parameters:
samples ('all' or list.) – A list of the sample names and/or grouped sample names to be plotted. All specified samples must be present in the CountFilter object. To average multiple replicates of the same condition, they can be grouped in an inner list. Example input: [[‘SAMPLE1A’, ‘SAMPLE1B’, ‘SAMPLE1C’], [‘SAMPLE2A’, ‘SAMPLE2B’, ‘SAMPLE2C’],’SAMPLE3’ , ‘SAMPLE6’]
scatter (bool (default=False)) – if True, adds a scatter-plot on top of the box-plot.
ylabel (str (default='Log10(RPM + 1)')) – the label of the Y axis.
title (str or 'auto' (default='auto')) – The title of the plot. If ‘auto’, a title will be generated automatically.
title_fontsize (float (default=30)) – determines the font size of the graph title.
label_fontsize (float (default=15)) – determines the font size of the X and Y axis labels.
tick_fontsize (float (default=10)) – determines the font size of the X and Y tick labels.
- Return type:
matplotlib Figure.
Example plot of enhanced_box_plot(add_scatter=True)
- filter_biotype_from_gtf(gtf_path: str | Path, biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
biotype (str or list of strings) – the biotypes which will not be filtered out.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- filter_biotype_from_ref_table(biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
- Parameters:
biotype (string or list of strings) – the biotypes which will not be filtered out.
ref – Name of the biotype reference file used to determine biotypes. Default is the path defined by the user in the settings.yaml file.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows whose biotype is 'protein_coding' >>> counts.filter_biotype_from_ref_table('protein_coding',ref='tests/test_files/biotype_ref_table_for_tests.csv') Filtered 9 features, leaving 13 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows whose biotype is 'protein_coding' or 'pseudogene' >>> counts.filter_biotype_from_ref_table(['protein_coding','pseudogene'],ref='tests/test_files/biotype_ref_table_for_tests.csv') Filtered 0 features, leaving 22 of the original 22 features. Filtered inplace.
- filter_by_attribute(attributes: str | List[str] = None, mode: Literal['union', 'intersection'] = 'union', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters features according to user-defined attributes from an Attribute Reference Table. When multiple attributes are given, filtering can be done in ‘union’ mode (where features that belong to at least one attribute are not filtered out), or in ‘intersection’ mode (where only features that belong to ALL attributes are not filtered out). To learn more about user-defined attributes and Attribute Reference Tables, read the user guide.
- Parameters:
attributes (string or list of strings, which are column titles in the user-defined Attribute Reference Table.) – attributes to filter by.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
ref (str or pathlib.Path (default='predefined')) – filename/path of the attribute reference table to be used as reference.
opposite (bool (default=False)) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to the attribute 'attribute1' >>> counts.filter_by_attribute('attribute1',ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 15 features, leaving 7 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to the attributes 'attribute1' OR 'attribute3' (union) >>> counts.filter_by_attribute(['attribute1','attribute3'],ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 14 features, leaving 8 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to both attributes 'attribute1' AND 'attribute3' (intersection) >>> counts.filter_by_attribute(['attribute1','attribute3'],mode='intersection', ... ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 19 features, leaving 3 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that DON'T belong to either 'attribute1','attribute3' or both >>> counts.filter_by_attribute(['attribute1','attribute3'],ref='tests/test_files/attr_ref_table_for_examples.csv', ... opposite=True) Filtered 8 features, leaving 14 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that DON'T belong to both 'attribute1' AND 'attribute3' >>> counts.filter_by_attribute(['attribute1','attribute3'],mode='intersection', ... ref='tests/test_files/attr_ref_table_for_examples.csv',opposite=True) Filtered 3 features, leaving 19 of the original 22 features. Filtered inplace.
- filter_by_go_annotations(go_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', propagate_annotations: bool = True, evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', opposite: bool = False, inplace: bool = True)
Filters genes according to GO annotations, keeping only genes that are annotated with a specific GO term. When multiple GO terms are given, filtering can be done in ‘union’ mode (where genes that belong to at least one GO term are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL GO terms are not filtered out).
- Parameters:
go_ids (str or list of str)
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :param propagate_annotations: determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/ :type propagate_annotations: ‘classic’, ‘elim’, ‘weight’, ‘all.m’, or ‘no’ (default=’elim’) :param evidence_types: only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or ‘any’ (default=’any’) :param excluded_evidence_types: annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type excluded_evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or None (default=None) :param databases: only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current FeatureSet object. If False, the function will return a new FeatureSet instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the FeatureSet object.
- filter_by_gtf_attribute(gtf_path: str | Path, attribute: Literal['gene_biotype', 'transcript_biotype', 'biotype', 'gene_name', 'gene_id', 'transcript_id', 'chromosome', 'source', 'strand'] | str = 'gene_biotype', value: str | List[str] = 'protein_coding', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters the features in the table by any attribute described in a GTF/GFF annotation file, keeping only features whose attribute matches one of the specified values (for example: keep only genes on a specific chromosome or strand, from a specific source, or of a specific biotype). This is a generalization of filter_biotype_from_gtf to any GTF/GFF attribute.
- Parameters:
gtf_path (str or Path) – Path to your GTF/GFF annotation file. The file should match the type of gene names/IDs you use in your table.
attribute (str (default='gene_biotype')) – name of the attribute to filter by. Standard column-9 attributes (such as ‘gene_biotype’, ‘gene_name’, or any custom key in your file) are supported, as well as the reserved names ‘chromosome’, ‘source’ and ‘strand’, which are read from the fixed columns of the annotation file.
value (str or list of strings) – the attribute value/values which will NOT be filtered out. For example, to keep only the features on chromosomes ‘chr1’ and ‘chr2’, set attribute=’chromosome’ and value=[‘chr1’, ‘chr2’].
feature_type ('gene' or 'transcript' (default='gene')) – determines whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- filter_by_kegg_annotations(kegg_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', opposite: bool = False, inplace: bool = True)
Filters genes according to KEGG pathways, keeping only genes that belong to specific KEGG pathway. When multiple KEGG IDs are given, filtering can be done in ‘union’ mode (where genes that belong to at least one pathway are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL pathways are not filtered out).
- Parameters:
kegg_ids (str or list of str) – the KEGG pathway IDs according to which the table will be filtered. An example for a legal KEGG pathway ID would be ‘path:cel04020’ for the C. elegans calcium signaling pathway.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the Filter object.
- filter_by_row_name(row_names: str | List[str], opposite: bool = False, inplace: bool = True)
Filter out specific rows from the table by their name (index).
- Parameters:
row_names (str or list of str) – list of row names to be removed from the table.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_by_row_sum(threshold: float = 5, opposite: bool = False, inplace: bool = True)
Removes features/rows whose sum is belove ‘threshold’.
- Parameters:
threshold (float) – The minimal sum a row should have in order not to be filtered out.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of CountFilter.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter('tests/test_files/counted.csv') >>> c.filter_by_row_sum(5) # remove all rows whose sum is <5 Filtered 4 features, leaving 18 of the original 22 features. Filtered inplace.
- filter_duplicate_ids(keep: Literal['first', 'last', 'neither'] = 'first', opposite: bool = False, inplace: bool = True)
Filter out rows with duplicate names/IDs (index).
- Parameters:
keep ('first', 'last', or 'neither' (default='first')) – determines which of the duplicates to keep for each group of duplicates. ‘first’ will keep the first duplicate found for each group; ‘last’ will keep the last; and ‘neither’ will remove all of the values in the group.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_low_reads(threshold: float = 5, n_samples: PositiveInt = 1, opposite: bool = False, inplace: bool = True)
Filter out features which are lowly-expressed in all columns, keeping only features with at least ‘threshold’ reads in at least ‘n_samples’ columns.
- Parameters:
threshold (float) – The minimal number of reads (counts, rpm, rpkm, tpm, etc) a feature should have in at least n_samples samples in order not to be filtered out.
n_samples (positive integer (default=1)) – the minimal number of samples a feature should have at least ‘threshold’ reads in in order not to be filtered out.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of CountFilter.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter('tests/test_files/counted.csv') >>> c.filter_low_reads(5) # remove all rows whose values in all columns are all <5 Filtered 6 features, leaving 16 of the original 22 features. Filtered inplace.
- filter_missing_values(columns: ColumnNames | Literal['all'] = 'all', opposite: bool = False, inplace: bool = True)
Remove all rows whose values in the specified columns are missing (NaN).
:param columns:name/names of the columns to check for missing values. :type opposite: bool :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/test_deseq_with_nan.csv') >>> filt_no_nan = filt.filter_missing_values(inplace=False) Filtered 3 features, leaving 25 of the original 28 features. Filtering result saved to new object. >>> filt_no_nan_basemean = filt.filter_missing_values(columns='baseMean', inplace=False) Filtered 1 features, leaving 27 of the original 28 features. Filtering result saved to new object. >>> filt_no_nan_basemean_pval = filt.filter_missing_values(columns=['baseMean','pvalue'], inplace=False) Filtered 2 features, leaving 26 of the original 28 features. Filtering result saved to new object.
- filter_percentile(percentile: Fraction, column: ColumnName, interpolate: 'nearest', 'higher', 'lower', 'midpoint', 'linear' = 'linear', opposite: bool = False, inplace: bool = True)
Removes all entries above the specified percentile in the specified column. For example, if the column were ‘pvalue’ and the percentile was 0.5, then all features whose pvalue is above the median pvalue will be filtered out.
- Parameters:
percentile (float between 0 and 1) – The percentile that all features above it will be filtered out.
column (str) – Name of the DataFrame column according to which the filtering will be performed.
interpolate ('nearest', 'higher', 'lower', 'midpoint' or 'linear' (default='linear')) – interpolation method to use when the desired quantile lies between two data points.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # keep only the rows whose value in the column 'log2FoldChange' is below the 75th percentile >>> d.filter_percentile(0.75,'log2FoldChange') Filtered 7 features, leaving 21 of the original 28 features. Filtered inplace.
>>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # keep only the rows vulse value in the column 'log2FoldChange' is above the 25th percentile >>> d.filter_percentile(0.25,'log2FoldChange',opposite=True) Filtered 7 features, leaving 21 of the original 28 features. Filtered inplace.
- filter_top_n(by: ColumnNames, n: PositiveInt = 100, ascending: bool | List[bool] = True, na_position: str = 'last', opposite: bool = False, inplace: bool = True)
Sort the rows by the values of specified column or columns, then keep only the top ‘n’ rows.
- Parameters:
by (name of column/columns (str/List[str])) – Names of the column or columns to sort and then filter by.
n (int) – How many features to keep in the Filter object.
ascending (bool or list of bools (default=True)) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of Filter.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only the 10 rows with the highest values in the columns 'cond1' >>> counts.filter_top_n(by='cond1',n=10, ascending=False) Filtered 12 features, leaving 10 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only the 10 rows which have the lowest values in the columns 'cond1' >>> # and then the highest values in the column 'cond2' >>> counts.filter_top_n(by=['cond1','cond2'],n=10, ascending=[True,False]) Filtered 12 features, leaving 10 of the original 22 features. Filtered inplace.
- find_paralogs_ensembl(organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True)
Find paralogs within the same species using the Ensembl database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
- Returns:
DataFrame describing all discovered paralog mappings.
- find_paralogs_panther(organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto')
Find paralogs within the same species using the PantherDB database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- Returns:
DataFrame describing all discovered paralog mappings.
- fname
- fold_change(numerator: ColumnNames, denominator: ColumnNames, numer_name: str = 'default', denom_name: str = 'default') FoldChangeFilter
Calculate the fold change between the numerator condition and the denominator condition, and return it as a FoldChangeFilter object.
- Parameters:
numerator (str, or list of strs) – the CountFilter columns to be used as the numerator. If multiple arguments are given in a list, they will be averaged.
denominator (str, or list of strs) – the CountFilter columns to be used as the denominator. If multiple arguments are given in a list, they will be averaged.
numer_name (str or 'default') – name to give the numerator condition. If ‘default’, the name will be generarated automatically from the names of numerator columns.
denom_name (str or 'default') – name to give the denominator condition. If ‘default’, the name will be generarated automatically from the names of denominator columns.
- Return type:
- Returns:
A new instance of FoldChangeFilter
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter('tests/test_files/counted_fold_change.csv') >>> # calculate the fold change of mean(cond1_rep1,cond1_rep2)/mean(cond2_rep1,cond_2rep2) >>> f = c.fold_change(['cond1_rep1','cond1_rep2'],['cond2_rep1','cond2_rep2']) >>> f.numerator "Mean of ['cond1_rep1', 'cond1_rep2']" >>> f.denominator "Mean of ['cond2_rep1', 'cond2_rep2']" >>> type(f) rnalysis.filtering.FoldChangeFilter
- classmethod from_folder(folder_path: str, save_csv: bool = False, fname: str = None, input_format: str = '.txt') CountFilter
Iterates over count .txt files in a given folder and combines them into a single CountFilter table. Can also save the count data table and the uncounted data table to .csv files.
- Parameters:
folder_path – str or pathlib.Path. Full path of the folder that contains individual htcount .txt files.
save_csv – bool. If True, the joint DataFrame of count data and uncounted data will be saved to two separate .csv files. The files will be saved in ‘folder_path’, and named according to the parameters ‘counted_fname’ for the count data, and ‘uncounted_fname’ for the uncounted data (unaligned, alignment not unique, etc).
fname – str. Name under which to save the combined count data table. Does not need to include the ‘.csv’ suffix.
input_format – the file format of the input files. Default is ‘.txt’.
- Returns:
an CountFilter object containing the combined count data from all individual htcount .txt files in the specified folder.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter.from_folder('tests/test_files/test_count_from_folder')
- classmethod from_folder_htseqcount(folder_path: str, norm_to_rpm: bool = False, save_csv: bool = False, counted_fname: str = None, uncounted_fname: str = None, input_format: str = '.txt') CountFilter
Iterates over HTSeq count .txt files in a given folder and combines them into a single CountFilter table. Can also save the count data table and the uncounted data table to .csv files, and normalize the CountFilter table to reads per million (RPM). Note that the saved data will always be count data, and not normalized data, regardless if the CountFilter table was normalized or not.
- Parameters:
folder_path – str or pathlib.Path. Full path of the folder that contains individual htcount .txt files.
norm_to_rpm – bool. If True, the CountFilter table will be automatically normalized to reads per million (RPM). If False (defualt), the CountFilter object will not be normalized, and will instead contain absolute count data (as in the original htcount .txt files). Note that if save_csv is True, the saved .csv fill will contain ABSOLUTE COUNT DATA, as in the original htcount .txt files, and NOT normalized data.
save_csv – bool. If True, the joint DataFrame of count data and uncounted data will be saved to two separate .csv files. The files will be saved in ‘folder_path’, and named according to the parameters ‘counted_fname’ for the count data, and ‘uncounted_fname’ for the uncounted data (unaligned, alignment not unique, etc).
counted_fname – str. Name under which to save the combined count data table. Does not need to include the ‘.csv’ suffix.
uncounted_fname – counted_fname: str. Name under which to save the combined uncounted data. Does not need to include the ‘.csv’ suffix.
input_format – the file format of the input files. Default is ‘.txt’.
- Returns:
an CountFilter object containing the combined count data from all individual htcount .txt files in the specified folder.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter.from_folder_htseqcount('tests/test_files/test_count_from_folder')
>>> c = filtering.CountFilter.from_folder_htseqcount('tests/test_files/test_count_from_folder', norm_to_rpm=True) # This will also normalize the CountFilter to reads-per-million (RPM).
Normalized 10 features. Normalized inplace.
>>> c = filtering.CountFilter.from_folder_htseqcount('tests/test_files/test_count_from_folder', save_csv=True, counted_fname='name_for_reads_csv_file', uncounted_fname='name_for_uncounted_reads_csv_file') # This will also save the counted reads and uncounted reads as separate .csv files
- head(n: PositiveInt = 5) DataFrame
Return the first n rows of the Filter object. See polars.DataFrame.head documentation.
- Parameters:
n (positive int, default 5) – Number of rows to show.
- Returns:
returns the first n rows of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> d.head() shape: (5, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬───────────┬─────────────┬─────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪═══════════╪═════════════╪═════════════╡ │ WBGene00000002 ┆ 6820.755327 ┆ 7.567762 ┆ 0.142257 ┆ 53.197692 ┆ 0.0 ┆ 0.0 │ │ WBGene00000003 ┆ 3049.62567 ┆ 9.138071 ┆ 0.245989 ┆ 37.148359 ┆ 4.6600e-302 ┆ 4.2800e-298 │ │ WBGene00000004 ┆ 1432.911791 ┆ 8.111737 ┆ 0.246801 ┆ 32.867521 ┆ 6.4000e-237 ┆ 3.9200e-233 │ │ WBGene00000005 ┆ 4028.154186 ┆ 6.534112 ┆ 0.202467 ┆ 32.272543 ┆ 1.7000e-228 ┆ 7.8000e-225 │ │ WBGene00000006 ┆ 1230.58524 ┆ 7.157428 ┆ 0.227948 ┆ 31.399325 ┆ 2.0700e-216 ┆ 7.5900e-213 │ └────────────────┴─────────────┴────────────────┴──────────┴───────────┴─────────────┴─────────────┘
>>> d.head(3) # return only the first 3 rows shape: (3, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬───────────┬─────────────┬─────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪═══════════╪═════════════╪═════════════╡ │ WBGene00000002 ┆ 6820.755327 ┆ 7.567762 ┆ 0.142257 ┆ 53.197692 ┆ 0.0 ┆ 0.0 │ │ WBGene00000003 ┆ 3049.62567 ┆ 9.138071 ┆ 0.245989 ┆ 37.148359 ┆ 4.6600e-302 ┆ 4.2800e-298 │ │ WBGene00000004 ┆ 1432.911791 ┆ 8.111737 ┆ 0.246801 ┆ 32.867521 ┆ 6.4000e-237 ┆ 3.9200e-233 │ └────────────────┴─────────────┴────────────────┴──────────┴───────────┴─────────────┴─────────────┘
- property index_set: set
Returns all of the features in the current DataFrame (which were not removed by previously used filter methods) as a set. if any duplicate features exist in the filter object (same WBGene appears more than once), the corresponding WBGene index will appear in the returned set ONLY ONCE.
- Returns:
A set of WBGene names.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> myset = counts.index_set >>> print(myset) {'WBGene00044022', 'WBGene00077504', 'WBGene00007079', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00077503', 'WBGene00007078', 'WBGene00007064', 'WBGene00077502', 'WBGene00044951', 'WBGene00007077', 'WBGene00007066', 'WBGene00007076', 'WBGene00014997', 'WBGene00043990', 'WBGene00007074', 'WBGene00043987', 'WBGene00007071', 'WBGene00043989', 'WBGene00043988', 'WBGene00007075'}
- property index_string: str
Returns a string of all feature indices in the current DataFrame, sorted by their current order in the FIlter object, and separated by newline.
- This includes all of the feature indices which were not filtered out by previously-used filter methods.
if any duplicate features exist in the filter object (same index appears more than once),
the corresponding index will appear in the returned string ONLY ONCE.
- Returns:
A string of WBGene indices separated by newlines (\n).
For example, “WBGene00000001\nWBGene00000003\nWBGene12345678”.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.sort(by='cond1',ascending=False) >>> mystring = counts.index_string >>> print(mystring) WBGene00007075 WBGene00043988 WBGene00043990 WBGene00007079 WBGene00007076 WBGene00043989 WBGene00007063 WBGene00007077 WBGene00007078 WBGene00007071 WBGene00007064 WBGene00007066 WBGene00007074 WBGene00043987 WBGene00007067 WBGene00014997 WBGene00044022 WBGene00077503 WBGene00077504 WBGene00077502 WBGene00007069 WBGene00044951
- intersection(*others: Filter | set, return_type: Literal['set', 'str'] = 'set', inplace: bool = False)
Keep only the features that exist in ALL of the given Filter objects/sets. Can be done either inplace on the first Filter object, or return a set/string of features.
- Parameters:
others (Filter or set objects.) – Objects to calculate intersection with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the intersecting features. If ‘str’, returns a string of the intersecting features, delimited by a comma.
inplace (bool (default=False)) – If True, the function will be applied in-place to the current Filter object. If False (default), the function will return a set/str that contains the intersecting indices.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that intersect between the given Filter objects/sets.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> # calculate intersection and return a set >>> d.intersection(a_set) {'WBGene00000002', 'WBGene00000003'}
# calculate intersection and filter in-place >>> d.intersection(a_set, inplace=True) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
- ma_plot(ref_column: Literal['auto'] | ColumnName = 'auto', columns: ColumnNames | Literal['all'] = 'all', split_plots: bool = False, title: str | Literal['auto'] = 'auto', title_fontsize: float = 20, label_fontsize: float | Literal['auto'] = 'auto', tick_fontsize: float = 12) List[Figure]
Generates M-A (log-ratio vs. log-intensity) plots for selected columns in the dataset. This plot is particularly useful for indicating whether a dataset is properly normalized.
- Parameters:
ref_column (name of a column or 'auto' (default='auto')) – the column to be used as reference for MA plot. If ‘auto’, then the reference column will be chosen automatically to be the column whose upper quartile is closest to the mean upper quartile.
columns (str or list of str) – A list of the column names to generate an MA plot for.
split_plots (bool (default=False)) – if True, each individual MA plot will be plotted in its own Figure. Otherwise, all MA plots will be plotted on the same Figure.
title (str or 'auto' (default='auto')) – The title of the plot. If ‘auto’, a title will be generated automatically.
title_fontsize (float (default=30)) – determines the font size of the graph title.
label_fontsize (float (default=15) :param tick_fontsize: determines the font size of the X and Y tick labels.) – determines the font size of the X and Y axis labels.
- Return type:
a list of matplotlib Figures.
- majority_vote_intersection(*others: Filter | set, majority_threshold: float = 0.5, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the features that appear in at least (majority_threhold * 100)% of the given Filter objects/sets. Majority-vote intersection with majority_threshold=0 is equivalent to Union. Majority-vote intersection with majority_threshold=1 is equivalent to Intersection.
- Parameters:
others (Filter or set objects.) – Objects to calculate intersection with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the intersecting WBGene indices. If ‘str’, returns a string of the intersecting indices, delimited by a comma.
majority_threshold (float (default=0.5)) – The threshold that determines what counts as majority. Features will be returned only if they appear in at least (majority_threshold * 100)% of the given Filter objects/sets.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that uphold majority vote intersection between two given Filter objects/sets.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> b_set = {'WBGene00000002','WBGene00000004'} >>> # calculate majority-vote intersection and return a set >>> d.majority_vote_intersection(a_set, b_set, majority_threshold=2/3) {'WBGene00000002', 'WBGene00000003', 'WBGene00000004'}
- map_orthologs_ensembl(map_to_organism: str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'], map_from_organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the Ensembl database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_orthoinspector(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the OrthoInspector database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_panther(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_least_diverged: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PantherDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_least_diverged (bool (default=True)) – if True (default), RNAlysis will only fetch ortholog mappings that were flagged as a ‘least diverged ortholog’ on the PantherDB database. You can read more about this flag on the PantherDB website: https://www.pantherdb.org/genes/
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_phylomedb(map_to_organism: str | int | Literal, map_from_organism: Literal['auto'] | str | int | Literal = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', consistency_score_threshold: Fraction = 0.5, filter_consistency_score: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PhylomeDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- param map_to_organism:
organism name or NCBI taxon ID of the target species for ortholog mapping.
- type map_to_organism:
str or int
- param map_from_organism:
organism name or NCBI taxon ID of the input genes’ source species.
- type map_from_organism:
str or int
- param gene_id_type:
the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- type gene_id_type:
str or ‘auto’ (default=’auto’)
- ` :param consistency_score_threshold: the minimum consistency score required for an ortholog mapping to be considered valid. Consistency scores are calculated by PhylomeDB and represent the confidence of the ortholog mapping. setting consistency_score_threshold to 0 will keep all mappings. You can read more about PhylomeDB consistency score on the PhylomeDB website: orthology.phylomedb.org/help
- type consistency_score_threshold:
float between 0 and 1 (default=0.5)
- param filter_consistency_score:
if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest consistency score.
- type filter_consistency_score:
bool (default=True)
- param non_unique_mode:
How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
- type non_unique_mode:
‘first’, ‘last’, ‘random’, or ‘none’ (default=’first’)
- param remove_unmapped_genes:
if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
- type remove_unmapped_genes:
bool (default=False)
- type inplace:
bool (default=True)
- param inplace:
If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- return:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- normalize_median_of_ratios(sample_grouping: GroupedColumns, reference_group: NonNegativeInt = 0, inplace: bool = True, return_scaling_factors: bool = False)
Normalizes the count matrix using the ‘Median of Ratios Normalization’ (MRN) method (Maza et al 2013). This normalization method uses information about the experimental condition of each sample. To calculate the Median of Ratios scaling factors, you first calculate the weighted mean expression of each gene within the replicates of each experimental condition. You then calculate per gene the ratio between each weighted mean in the experimental condition and those of the reference condition. You then pick the median ratio for each experimental condition, and calculate the scaling factor for each sample by multiplying it with the sample’s total number of reads. Finally, the scaling factors are adjusted, for symmetry, so that they multiply to 1.
- Parameters:
sample_grouping (nested list of column names) – grouping of the samples into conditions. Each grouping should containg all replicates of the same condition.
reference_group (int (default=0)) – the index of the sample group to be used as the reference condition. Must be an integer between 0 and the number of sample groups -1.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
return_scaling_factors (bool (default=False)) – if True, return a DataFrame containing the calculated scaling factors.
- Returns:
If inplace is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter("tests/test_files/counted.csv") >>> c.normalize_median_of_ratios([['cond1','cond2'],['cond3','cond4']])
Normalized 22 features. Normalized inplace.
- normalize_rle(inplace: bool = True, return_scaling_factors: bool = False)
Normalizes the count matrix using the ‘Relative Log Expression’ (RLE) method (Anders and Huber 2010). This is the default normalization method used by R’s DESeq2. To calculate the Relative Log Expression scaling factors, you first generate a pseudo-sample by calculating the geometric mean expression of each gene across samples. You then calculate the gene-wise ratio of expression between each sample and the pseudo-sample. You then pick the median ratio within each sample as the scaling factor for that sample.
- Parameters:
inplace (bool (default=True)) – If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
return_scaling_factors (bool (default=False)) – if True, return a DataFrame containing the calculated scaling factors.
- Returns:
If inplace is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter("tests/test_files/counted.csv") >>> c.normalize_rle()
Normalized 22 features. Normalized inplace.
- normalize_tmm(log_ratio_trim: float = 0.3, sum_trim: float = 0.05, a_cutoff: float | None = -10000000000, ref_column: Literal['auto'] | ColumnName = 'auto', inplace: bool = True, return_scaling_factors: bool = False)
Normalizes the count matrix using the ‘trimmed mean of M values’ (TMM) method (Robinson and Oshlack 2010). This is the default normalization method used by R’s edgeR. To calculate the Trimmed Mean of M Values scaling factors, you first calculate the M-values of each gene between each sample and the reference sample (log2 of each sample Minus log2 of the reference sample), and the A-values of each gene between each sample and the reference sample (log2 of each sample Added to log2 of the reference sample). You then trim out genes with extreme values that are likely to be differentially expressed or non-indicative, by trimming the top and bottom X% of M-values, the top and bottom Y% of A-values, all A-values which are smaller than the specified cutuff, and all genes with 0 reads (to avoid log2 values of inf or -inf). Next, a weighted mean is calculated on the filtered M-values, with the weights being an inverse of an approximation of variance of each gene, which gives out the scaling factors for each sample. Finally, the scaling factors are adjusted, for symmetry, so that they multiply to 1.
- Parameters:
log_ratio_trim (float between 0 and 0.5 (default=0.3)) – the fraction of M-values that should be trimmed from each direction (top and bottom X%).
sum_trim (float between 0 and 0.5 (default=0.05)) – the fraction of A-values that should be trimmed from each direction (top and bottom Y%).
a_cutoff (float or None (default = -1e10)) – a lower bound on the A-values that should be included in the trimmed mean. If set to None, no lower bound will be used.
ref_column (name of a column or 'auto' (default='auto')) – the column to be used as reference for normalization. If ‘auto’, then the reference column will be chosen automatically to be the column whose upper quartile is closest to the mean upper quartile.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
return_scaling_factors (bool (default=False)) – if True, return a DataFrame containing the calculated scaling factors.
- Returns:
If inplace is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter("tests/test_files/counted.csv") >>> c.normalize_tmm()
Normalized 22 features. Normalized inplace.
- normalize_to_quantile(quantile: Fraction = 0.75, inplace: bool = True, return_scaling_factors: bool = False)
Normalizes the count matrix using the quantile method, generalized from Bullard et al 2010. This is the default normalization method used by R’s Limma. To calculate the Quantile Method scaling factors, you first calculate the given quantile of gene expression within each sample, excluding genes that have 0 reads in all samples. You then divide those quantile values by the total number of reads in each sample, which yields the scaling factors for each sample.
- Parameters:
quantile (float between 0 and 1 (default=0.75)) – the quantile from which scaling factors will be calculated.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
return_scaling_factors (bool (default=False)) – if True, return a DataFrame containing the calculated scaling factors.
- Returns:
If inplace is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter("tests/test_files/counted.csv") >>> c.normalize_to_quantile(0.75)
Normalized 22 features. Normalized inplace.
- normalize_to_rpkm(gtf_file: str | Path, feature_type: Literal['gene', 'transcript'] = 'gene', method: Literal['mean', 'median', 'max', 'min', 'geometric_mean', 'merged_exons'] = 'mean', inplace: bool = True, return_scaling_factors: bool = False)
Normalizes the count matrix to Reads Per Kilobase Million (RPKM). Divides each column in the count matrix by (total reads)*(gene length / 1000)*10^-6.
- Parameters:
gtf_file – Path to a GTF/GFF3 annotation file. This file will be used to determine the length of each gene/transcript. The gene/transcript names in this annotation file should match the ones in count matrix. :type gtf_file: str or Path :param feature_type: the type of features in your count matrix. if feature_type is ‘transcript’, lengths will be calculated per-transcript, and the ‘method’ parameter is ignored. Otherwise, lengths will be aggregated per gene according to the method specified in the ‘method’ parameter. :type feature_type: ‘gene’ or ‘transcript’ (default=’gene’) :param method: if feature_type=’gene’, this determines the aggregation method to calculate gene lengths. ‘mean’, ‘median’, ‘min’, and ‘max’ will calculate the mean/median/min/max of all transcripts’ lengths of the given gene. ‘geometric_mean’ will calculate the goemetric mean of all transcripts’ lengths of the given gene. ‘merged_exons’ will calculate the total lengths of all exons of a gene across all of its transcripts, while counting overlapping exons/regions exactly once. :type method: ‘mean’, ‘median’, ‘min’, ‘max’, ‘geometric_mean’, or ‘merged_exons’ (deafult=’mean’) :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected. :param return_scaling_factors: if True, return a DataFrame containing the calculated scaling factors. :type return_scaling_factors: bool (default=False) :return: If inplace is False, returns a new instance of the Filter object.
- normalize_to_rpm(inplace: bool = True, return_scaling_factors: bool = False)
Normalizes the count matrix to Reads Per Million (RPM). Divides each column in the count matrix by (total reads)*10^-6 .
- Parameters:
inplace (bool (default=True)) – If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
return_scaling_factors (bool (default=False)) – if True, return a DataFrame containing the calculated scaling factors.
- Returns:
If inplace is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter("tests/test_files/counted.csv") >>> c.normalize_to_rpm()
Normalized 22 features. Normalized inplace.
- normalize_to_rpm_htseqcount(special_counter_fname: str | Path, inplace: bool = True, return_scaling_factors: bool = False)
Normalizes the count matrix to Reads Per Million (RPM). Uses a table of feature counts (ambiguous, no feature, not aligned, etc) from HTSeq-count’s output. Divides each column in the CountFilter object by (total reads + ambiguous + no feature)*10^-6 .
- Parameters:
special_counter_fname – the .csv file which contains feature information about the RNA library (ambiguous, no feature, not aligned, etc).
inplace (bool (default=True)) – If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
return_scaling_factors (bool (default=False)) – if True, return a DataFrame containing the calculated scaling factors.
- Returns:
If inplace is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter("tests/test_files/counted.csv") >>> c.normalize_to_rpm_htseqcount("tests/test_files/uncounted.csv")
Normalized 22 features. Normalized inplace.
- normalize_to_tpm(gtf_file: str | Path, feature_type: Literal['gene', 'transcript'] = 'gene', method: Literal['mean', 'median', 'max', 'min', 'geometric_mean', 'merged_exons'] = 'mean', inplace: bool = True, return_scaling_factors: bool = False)
Normalizes the count matrix to Transcripts Per Million (TPM). First, normalizes each gene to Reads Per Kilobase (RPK) by dividing each gene in the count matrix by its length in Kbp (gene length / 1000). Then, divides each column in the RPK matrix by (total RPK in column)*10^-6. This calculation is similar to that of Reads Per Kilobase Million (RPKM), but in the opposite order: the “per million” normalization factors are calculated after normalizing to gene lengths, not before.
- Parameters:
gtf_file – Path to a GTF/GFF3 annotation file. This file will be used to determine the length of each gene/transcript. The gene/transcript names in this annotation file should match the ones in count matrix. :type gtf_file: str or Path :param feature_type: the type of features in your count matrix. if feature_type is ‘transcript’, lengths will be calculated per-transcript, and the ‘method’ parameter is ignored. Otherwise, lengths will be aggregated per gene according to the method specified in the ‘method’ parameter. :type feature_type: ‘gene’ or ‘transcript’ (default=’gene’) :param method: if feature_type=’gene’, this determines the aggregation method to calculate gene lengths. ‘mean’, ‘median’, ‘min’, and ‘max’ will calculate the mean/median/min/max of all transcripts’ lengths of the given gene. ‘geometric_mean’ will calculate the goemetric mean of all transcripts’ lengths of the given gene. ‘merged_exons’ will calculate the total lengths of all exons of a gene across all of its transcripts, while counting overlapping exons/regions exactly once. :type method: ‘mean’, ‘median’, ‘min’, ‘max’, ‘geometric_mean’, or ‘merged_exons’ (deafult=’mean’) :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected. :param return_scaling_factors: if True, return a DataFrame containing the calculated scaling factors. :type return_scaling_factors: bool (default=False) :return: If inplace is False, returns a new instance of the Filter object.
- normalize_with_scaling_factors(scaling_factor_fname: str | Path, inplace: bool = True)
Normalizes the reads in the CountFilter using pre-calculated scaling factors. Receives a table of sample names and their corresponding scaling factors, and divides each column in the CountFilter by the corresponding scaling factor.
- Parameters:
scaling_factor_fname (str or pathlib.Path) – the .csv file which contains scaling factors for the different libraries.
inplace – If True (default), filtering will be applied to the current CountFilter object. If False, the function will return a new CountFilter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter("tests/test_files/counted.csv") >>> c.normalize_with_scaling_factors("tests/test_files/scaling_factors.csv")
Normalized 22 features. Normalized inplace.
- number_filters(column: ColumnName, operator: Literal['greater than', 'equals', 'lesser than', 'abs greater than'], value: float, opposite: bool = False, inplace: bool = True)
Applay a number filter (greater than, equal, lesser than) on a particular column in the Filter object.
- Parameters:
column (str) – name of the column to filter by
operator (str: 'gt' / 'greater than' / '>', 'eq' / 'equals' / '=', 'lt' / 'lesser than' / '<') – the operator to filter the column by (greater than, equal or lesser than)
value (float) – the value to filter by
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','gt',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
>>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','greater than',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
>>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','>',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
- pairplot(samples: GroupedColumns | Literal['all'] = 'all', log2: bool = True, show_corr: bool = True, title: str | Literal['auto'] = 'auto', title_fontsize: float = 30, label_fontsize: float = 16, tick_fontsize: float = 12) Figure
Plot pairwise relationships in the dataset. Can plot both single samples and average multiple replicates. For more information see the documentation of seaborn.pairplot.
- Parameters:
samples ('all', list, or nested list.) – A list of the sample names and/or grouped sample names to be included in the pairplot. All specified samples must be present in the CountFilter object. To average multiple replicates of the same condition, they can be grouped in an inner list. Example input: [[‘SAMPLE1A’, ‘SAMPLE1B’, ‘SAMPLE1C’], [‘SAMPLE2A’, ‘SAMPLE2B’, ‘SAMPLE2C’],’SAMPLE3’ , ‘SAMPLE6’]
log2 (bool (default=True)) – if True, the pairplot will be calculated with log2 of the DataFrame (pseudocount+1 added), and not with the raw data. If False, the pairplot will be calculated with the raw data.
show_corr (bool (default=True)) – if True, shows the Spearman correlation coefficient (R) between each pair of samples/groups.
title (str or 'auto' (default='auto')) – The title of the plot. If ‘auto’, a title will be generated automatically.
title_fontsize (float (default=30)) – determines the font size of the graph title.
label_fontsize (float (default=15) :param tick_fontsize: determines the font size of the X and Y tick labels.) – determines the font size of the X and Y axis labels.
- Returns:
A matplotlib Figure.
Example plot of pairplot()
- pca(samples: GroupedColumns | Literal['all'] = 'all', n_components: PositiveInt = 3, power_transform: bool = True, labels: bool = True, title: str | Literal['auto'] = 'auto', title_fontsize: float = 20, label_fontsize: float = 16, tick_fontsize: float = 12, proportional_axes: bool = False, plot_grid: bool = True, legend: List[str] | None = None) Tuple[PCA, List[Figure]]
Performs Principal Component Analysis (PCA), visualizing the principal components that explain the most variance between the different samples. The function will standardize the data prior to PCA, and then plot the requested number of pairwise PCA projections.
- Parameters:
samples ('all' or list.) – A list of the sample names and/or grouped sample names to be plotted. All specified samples must be present in the CountFilter object. To draw multiple replicates of the same condition in the same color, they can be grouped in an inner list. Example input: [[‘SAMPLE1A’, ‘SAMPLE1B’, ‘SAMPLE1C’], [‘SAMPLE2A’, ‘SAMPLE2B’, ‘SAMPLE2C’],’SAMPLE3’ , ‘SAMPLE6’]
n_components (int >=2 (default=3)) – number of Principal Components to plot (minimum is 2). RNAlysis will generate a pair-wise scatter plot between every pair of Principal Components.
labels (bool (default=True)) – if True, RNAlysis will display labels with the sample names next to each sample on the graph.
power_transform (bool (default=True)) – if True, RNAlysis will apply a power transform (Box-Cox) to the data prior to standartization and principal component analysis.
title (str or 'auto' (default='auto')) – The title of the plot. If ‘auto’, a title will be generated automatically.
title_fontsize (float (default=30)) – determines the font size of the graph title.
label_fontsize (float (default=15)) – determines the font size of the X and Y axis labels.
tick_fontsize (float (default=10)) – determines the font size of the X and Y tick labels, and the sample name labels. .
proportional_axes (bool (default=False)) – if True, the dimensions of the PCA plots will be proportional to the percentage of variance explained by each principal component.
plot_grid (bool (default=True)) – if True, will draw a grid on the PCA plot.
legend (list of str, or None (default=None)) – if enabled, display a legend on the PCA plot. Each entry in the ‘legend’ parameter corresponds to one group of samples (one color on the graph), as defined by the parameter ‘samples’
- Returns:
A tuple whose first element is an sklearn.decomposition.pca object, and second element is a list of matplotlib.axis objects.
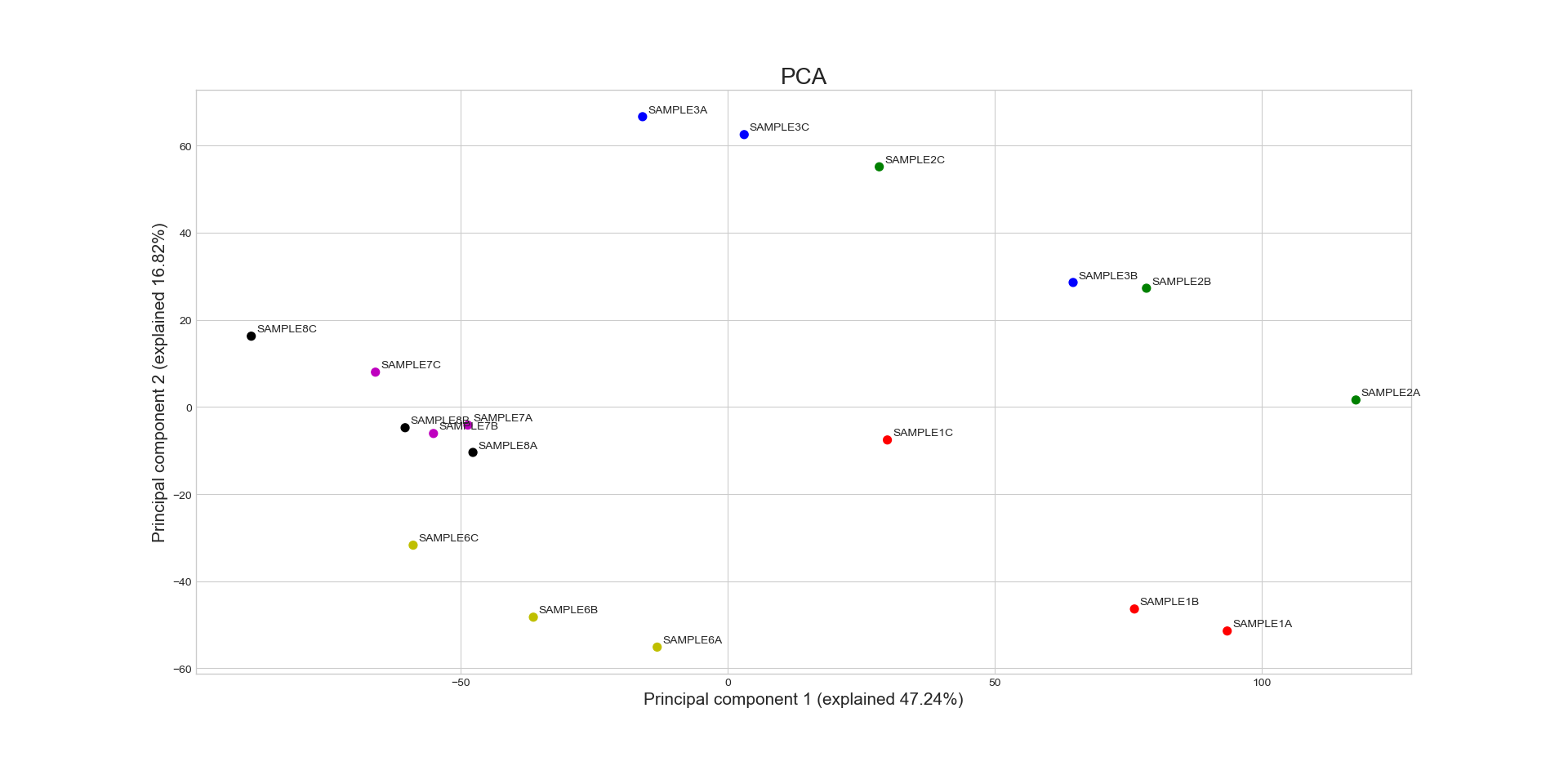
Example plot of pca()
- plot_expression(features: List[str] | str, samples: GroupedColumns | Literal['all'] = 'all', avg_function: Literal['mean', 'median', 'geometric_mean'] = 'mean', spread_function: Literal['sem', 'std', 'gstd', 'gsem', 'iqr', 'range'] = 'sem', bar_colors: ColorList = 'deepskyblue', edge_color: Color = 'black', scatter_color: Color = 'grey', count_unit: str = 'Normalized reads', split_plots: bool = False, jitter: Fraction = 0, group_names: List[str] | None = None, log_scale: bool = False) Figure
Plot the average expression and spread of the specified features under the specified conditions. :type features: str or list of strings :param features: the feature/features to plot expression for. :param samples: A list of the sample names and/or grouped sample names to be plotted. All specified samples must be present in the CountFilter object. To average multiple replicates of the same condition, they can be grouped in an inner list. Example input: [[‘SAMPLE1A’, ‘SAMPLE1B’, ‘SAMPLE1C’], [‘SAMPLE2A’, ‘SAMPLE2B’, ‘SAMPLE2C’],’SAMPLE3’ , ‘SAMPLE6’] :param avg_function: The function used to calculate the average expression value for each condition. :type avg_function: ‘mean’, ‘median’, or ‘geometric_mean’ (default=’mean’) :param spread_function: The function used to calculate the error bars of expression values for each condition. :type spread_function: ‘sem’, ‘std’, ‘gstd’, ‘gsem’, ‘iqr’, or ‘range’ (default=’sem’) :param bar_colors: The color or list of colors to use for the bars in the plot. :type bar_colors: str or list of color strings (default=’deepskyblue’) :param edge_color: The color of the edges around the bars. :type edge_color: str (default=’black’) :param scatter_color: The color of the scatter points representing individual samples. :type scatter_color: str (default=’grey’) :type count_unit: str (default=’Reads per million’) :param count_unit: The unit of the count data. Will be displayed in the y axis. :type split_plots: bool (default=False) :param split_plots: if True, each gene will be plotted in its own Figure. Otherwise, all genes will be plotted in the same Figure. :type jitter: float (default=0) :param jitter: The amount of jitter to apply to the scatter points. This can help visualize overlapping points. :type group_names: list of strings or None (default=None) :param group_names: Optionally, specify the names of the groups in the plot. If None, the names of the samples will be used. :type log_scale: bool (default=False) :param log_scale: If True, the y-axis will be displayed in logarithmic scale.
Example plot of plot_expression()
- print_features()
Print the feature indices in the Filter object, sorted by their current order in the FIlter object, and separated by newline.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.sort(by='cond1',ascending=False) >>> counts.print_features() WBGene00007075 WBGene00043988 WBGene00043990 WBGene00007079 WBGene00007076 WBGene00043989 WBGene00007063 WBGene00007077 WBGene00007078 WBGene00007071 WBGene00007064 WBGene00007066 WBGene00007074 WBGene00043987 WBGene00007067 WBGene00014997 WBGene00044022 WBGene00077503 WBGene00077504 WBGene00077502 WBGene00007069 WBGene00044951
- save_csv(alt_filename: None | str | Path = None)
Saves the current filtered data to a .csv file.
- Parameters:
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- save_parquet(alt_filename: None | str | Path = None)
Saves the current filtered data to a .parquet file.
- Parameters:
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- save_table(suffix: Literal['.csv', '.tsv', '.parquet'] = '.csv', alt_filename: None | str | Path = None)
Save the current filtered data table.
- Parameters:
suffix ('.csv', '.tsv', or '.parquet' (default='.csv')) – the file suffix
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- scatter_sample_vs_sample(sample1: ColumnNames, sample2: ColumnNames, xlabel: str | Literal['auto'] = 'auto', ylabel: str | Literal['auto'] = 'auto', title: str | Literal['auto'] = 'auto', title_fontsize: float = 20, label_fontsize: float = 16, tick_fontsize: float = 12, annotation_fontsize: float = 10, highlight: Sequence[str] | None = None, point_color: Color = '#6d7178', highlight_color: Color = '#00aaff', opacity: Fraction = 0.65, point_size: float = 10, interactive: bool = True, show_cursor: bool = False) Figure
Generate a scatter plot where every dot is a feature, the x value is log10 of reads (counts, RPM, RPKM, TPM, etc) in sample1, the y value is log10 of reads in sample2. If the plot is generated in interactive mode, data points can be labeled by clicking on them.
- Parameters:
sample1 (string or list of strings) – Name of the first sample from the CountFilter object. If sample1 is a list, they will be avarged as replicates.
sample2 (string or list of strings) – Name of the second sample from the CountFilter object. If sample2 is a list, they will be averaged as replicates.
xlabel (str or 'auto') – optional. If not specified, sample1 will be used as xlabel.
ylabel (str or 'auto') – optional. If not specified, sample2 will be used as ylabel.
title (str or 'auto') – optional. If not specified, a title will be generated automatically.
title_fontsize (float (default=30)) – determines the font size of the graph title.
label_fontsize (float (default=15)) – determines the font size of the X and Y axis labels.
tick_fontsize (float (default=10)) – determines the font size of the X and Y tick labels.
annotation_fontsize (float (default=10)) – determines the font size of the point annotations created in interactive mode.
highlight (Filter object or iterable of strings) – If specified, the points in the scatter corresponding to the names/features in ‘highlight’ will be highlighted in red.
point_color (str or tuple of (int, int, int) (default='#6d7178')) – color of the points in the scatter plot.
highlight_color (str or tuple of (int, int, int) (default='#00aaff')) – color of the highlighted points in the scatter plot.
opacity (determines the opacity of the points in the scatter plot. 0 indicates completely transparent, while 1 indicates completely opaque.) – float between 0 and 1 (default=0.65)
point_size (float (default=10)) – determines the size of the points in the scatter plot
interactive (bool (default=True)) – if True, turns on interactive mode. While in interactive mode, you can click on a data point to label it with its gene name/ID, or click on a labeled data point to unlabel it.
show_cursor (bool (default=False)) – if True, show the cursor position on the plot during interactive mode
- Returns:
a matplotlib axis object.
Example plot of scatter_sample_vs_sample()
- sort(by: str | List[str], ascending: bool | List[bool] = True, na_position: str = 'last', inplace: bool = True)
Sort the rows by the values of specified column or columns.
- Parameters:
by (str or list of str) – Names of the column or columns to sort by.
ascending (bool or list of bool (default=True)) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
inplace (bool (default=True)) – If True, perform operation in-place. Otherwise, returns a sorted copy of the Filter object without modifying the original.
- Returns:
None if inplace=True, a sorted Filter object otherwise.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.head() cond1 cond2 cond3 cond4 WBGene00007063 633 451 365 388 WBGene00007064 60 57 20 23 WBGene00044951 0 0 0 1 WBGene00007066 55 266 46 39 WBGene00007067 15 13 1 0 >>> counts.sort(by='cond1',ascending=True) >>> counts.head() cond1 cond2 cond3 cond4 WBGene00044951 0 0 0 1 WBGene00077504 0 0 0 0 WBGene00007069 0 2 1 0 WBGene00077502 0 0 0 0 WBGene00077503 1 4 2 0
- sort_by_principal_component(component: PositiveInt, ascending: bool = True, power_transform: bool = True, inplace: bool = True)
Performs Principal Component Analysis (PCA), and sort the table based on the contribution (loadings) of genes to a specific Principal Component. This type of analysis can help you understand which genes contribute the most to each principal component, particularly using single-list enrichment analysis. .
- Parameters:
component (positive int) – the Principal Component the table should be sorted by.
ascending (bool (default=Trle)) – Sort order: ascending (negative loadings at the top of the list) versus descending (positive loadings at the top of the list).
power_transform (bool (default=True)) – if True, RNAlysis will apply a power transform (Box-Cox) to the data prior to standartization and principal component analysis.
inplace (bool (default=True)) – If True, perform the operation in-place. Otherwise, returns a sorted copy of the Filter object without modifying the original.
- Returns:
None if inplace=True, a sorted Filter object otherwise.
- split_by_attribute(attributes: str | List[str], ref: str | Path | Literal['predefined'] = 'predefined') tuple
Splits the features in the Filter object into multiple Filter objects, each corresponding to one of the specified Attribute Reference Table attributes. Each new Filter object will contain only features that belong to its Attribute Reference Table attribute.
- Parameters:
attributes (list of strings) – list of attribute names from the Attribute Reference Table to filter by.
ref – filename/path of the reference table to be used as reference.
- Return type:
Tuple[filtering.Filter]
- Returns:
A tuple of Filter objects, each containing only features that match one Attribute Reference Table attribute; the Filter objects are returned in the same order the attributes were given in.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> attribute1,attribute2 = counts.split_by_attribute(['attribute1','attribute2'], ... ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 15 features, leaving 7 of the original 22 features. Filtering result saved to new object. Filtered 20 features, leaving 2 of the original 22 features. Filtering result saved to new object.
- split_by_percentile(percentile: Fraction, column: ColumnName, interpolate: 'nearest', 'higher', 'lower', 'midpoint', 'linear' = 'linear') tuple
Splits the features in the Filter object into two non-overlapping Filter objects: one containing features below the specified percentile in the specfieid column, and the other containing features about the specified percentile in the specified column.
- Parameters:
percentile (float between 0 and 1) – The percentile that all features above it will be filtered out.
column (str) – Name of the DataFrame column according to which the filtering will be performed.
- Return type:
Tuple[filtering.Filter, filtering.Filter]
- Returns:
a tuple of two Filter objects: the first contains all of the features below the specified percentile, and the second contains all of the features above and equal to the specified percentile.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> below, above = d.split_by_percentile(0.75,'log2FoldChange') Filtered 7 features, leaving 21 of the original 28 features. Filtering result saved to new object. Filtered 21 features, leaving 7 of the original 28 features. Filtering result saved to new object.
- split_by_principal_components(components: PositiveInt | List[PositiveInt], gene_fraction: Fraction = 0.1, power_transform: bool = True) Tuple[CountFilter, CountFilter] | Tuple[Tuple[CountFilter, CountFilter], ...]
Performs Principal Component Analysis (PCA), and split the table based on the contribution (loadings) of genes to specific Principal Components. For each Principal Component specified, RNAlysis will find the X% most influential genes on the Principal Component based on their loadings (where X is gene_fraction), (X/2)% from the top and (X/2)% from the bottom. This type of analysis can help you understand which genes contribute the most to each principal component.
- Parameters:
components (int or list of integers) – the Principal Components the table should be filtered by. Each Principal Component will be analyzed separately.
gene_fraction (float between 0 and 1 (default=0.1)) – the total fraction of top influential genes that will be returned. For example, if gene_fraction=0.1, RNAlysis will return the top and bottom 5% of genes based on their loadings for any principal component.
power_transform (bool (default=True)) – if True, RNAlysis will apply a power transform (Box-Cox) to the data prior to standartization and principal component analysis.
- split_by_reads(threshold: float = 5) tuple
Splits the features in the CountFilter object into two non-overlapping CountFilter objects, based on their maximum expression level. The first object will contain only highly-expressed features (which have reads over the specified threshold in at least one sample). The second object will contain only lowly-expressed features (which have reads below the specified threshold in all samples).
- Parameters:
threshold (float (default=5)) – The minimal number of reads (counts, RPM, RPKM, TPM etc) a feature needs to have in at least one sample in order to be included in the “highly expressed” object and no the “lowly expressed” object.
- Return type:
- Returns:
A tuple containing two CountFilter objects: the first has only highly-expressed features, and the second has only lowly-expressed features.
- Examples:
>>> from rnalysis import filtering >>> c = filtering.CountFilter('tests/test_files/counted.csv') >>> low_expression, high_expression = c.split_by_reads(5) Filtered 6 features, leaving 16 of the original 22 features. Filtering result saved to new object. Filtered 16 features, leaving 6 of the original 22 features. Filtering result saved to new object.
- split_clicom(*parameter_dicts: dict, replicate_grouping: GroupedColumns | Literal['ungrouped'] = 'ungrouped', power_transform: bool | Tuple[bool, bool] = True, evidence_threshold: Fraction = 0.6666666666666666, cluster_unclustered_features: bool = False, min_cluster_size: PositiveInt = 15, plot_style: Literal['all', 'std_area', 'std_bar'] = 'all', split_plots: bool = False, parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) Tuple[CountFilter, ...]
Clusters the features in the CountFilter object using the modified CLICOM ensemble clustering algorithm (Mimaroglu and Yagci 2012), and then splits those features into multiple non-overlapping CountFilter objects, based on the clustering result. The CLICOM algorithm incorporates the results of multiple clustering solutions, which can come from different clustering algorithms with differing clustering parameters, and uses these clustering solutions to create a combined clustering solution. Due to the nature of CLICOM, the number of clusters the data will be divided into is determined automatically. This modified version of the CLICOM algorithm can also classify features as noise, which does not belong in any discovered cluster.
- Parameters:
replicate_grouping (nested list of strings or 'ungrouped' (default='ungrouped')) – Allows you to group your data into replicates. Each replicate will be clustered separately, and used as its own clustering setup. This can minimize the influence of batch effects on the clustering results, and take advantage of repeated measures data to improve the accuracy of your clustering. If `replicate_grouping`=’ungrouped’, the data will be clustered normally as if no replicate data is available. To read more about the theory behind this, see the following publication: https://doi.org/10.1093/bib/bbs057
power_transform (True, False, or (True, False) (default=True)) – if True, RNAlysis will apply a power transform (Box-Cox) to the data prior to clustering. If both True and False are supplied, RNAlysis will run the initial clustering setups twice: once with a power transform, and once without.
evidence_threshold (float between 0 and 1 (default=2/3)) – determines whether each pair of features can be reliably clustered together. For example, if evidence_threshold=0.5, a pair of features is considered reliably clustered together if they were clustered together in at least 50% of the tested clustering solutions.
cluster_unclustered_features (bool (default=False)) – if True, RNAlysis will force every feature to be part of a cluster, even if they were not initially determined to reliably belong to any of the discovered clusters. Larger values will lead to fewer clusters, with more features classified as noise.
min_cluster_size (int (default=15)) – the minimum size of clusters the algorithm will seek. Larger values will lead to fewer clusters, with more features classified as noise.
parameter_dicts – multiple dictionaries, each corresponding to a clustering setup to be run. Each dictionary must contain a ‘method’ field with a clustering method supported by RNAlysis (‘k-means’, ‘k-medoids’, ‘hierarchical’, or ‘hdbscan’). The other fields of the dictionary should contain your preferred values for each of the clustering algorithm’s parameters. Yoy can specify a list of values for each of those parameters, and then RNAlysis will run the clustering algorithm with all legal combinations of parameters you specified. For example, {‘method’:’k-medoids’, ‘n_clusters’:[3,5], ‘metric’:[‘Euclidean’, ‘cosine’]} will run the K-Medoids algorithm four times with the following parameter combinations: (n_clusters=3,metric=’Euclidean’), (n_clusters=5, metric=’Euclidean’), (n_clusters=3, metric=’cosine’), (n_clusters=5, metric=’cosine’).
plot_style ('all', 'std_area', or 'std_bar' (default='all')) – determines the visual style of the cluster expression plot.
split_plots (bool (default=False)) – if True, each discovered cluster will be plotted on its own. Otherwise, all clusters will be plotted in the same Figure.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Returns:
returns a tuple of CountFilter objects, each corresponding to a discovered cluster.
- Examples:
>>> from rnalysis import filtering >>> dev_stages = filtering.CountFilter('tests/test_files/elegans_developmental_stages.tsv') >>> dev_stages.filter_low_reads(100) Filtered 44072 features, leaving 2326 of the original 46398 features. Filtered inplace. >>> clusters = dev_stages.split_clicom( ... {'method': 'hdbscan', 'min_cluster_size': [50, 75, 140], 'metric': ['ys1', 'yr1', 'spearman']}, ... {'method': 'hierarchical', 'n_clusters': [7, 12], 'metric': ['Euclidean', 'jackknife', 'yr1'], ... 'linkage': ['average', 'ward']}, {'method': 'kmedoids', 'n_clusters': [7, 16], 'metric': 'spearman'}, ... power_transform=True, evidence_threshold=0.5, min_cluster_size=40) Found 19 legal clustering setups. Running clustering setups: 100%|██████████| 19/19 [00:12<00:00, 1.49 setup/s] Generating cluster similarity matrix: 100%|██████████| [00:32<00:00, 651.06it/s] Finding cliques: 100%|██████████| 42436/42436 [00:00<00:00, 61385.87it/s] Done Found 15 clusters of average size 153.60. Number of unclustered genes is 22, which are 0.95% of the genes. Filtered 1864 features, leaving 462 of the original 2326 features. Filtering result saved to new object. Filtered 2115 features, leaving 211 of the original 2326 features. Filtering result saved to new object. Filtered 2122 features, leaving 204 of the original 2326 features. Filtering result saved to new object. Filtered 2123 features, leaving 203 of the original 2326 features. Filtering result saved to new object. Filtered 2128 features, leaving 198 of the original 2326 features. Filtering result saved to new object. Filtered 2167 features, leaving 159 of the original 2326 features. Filtering result saved to new object. Filtered 2179 features, leaving 147 of the original 2326 features. Filtering result saved to new object. Filtered 2200 features, leaving 126 of the original 2326 features. Filtering result saved to new object. Filtered 2204 features, leaving 122 of the original 2326 features. Filtering result saved to new object. Filtered 2229 features, leaving 97 of the original 2326 features. Filtering result saved to new object. Filtered 2234 features, leaving 92 of the original 2326 features. Filtering result saved to new object. Filtered 2238 features, leaving 88 of the original 2326 features. Filtering result saved to new object. Filtered 2241 features, leaving 85 of the original 2326 features. Filtering result saved to new object. Filtered 2263 features, leaving 63 of the original 2326 features. Filtering result saved to new object. Filtered 2279 features, leaving 47 of the original 2326 features. Filtering result saved to new object.
Example plot of split_clicom(plot_style=’all’)
Example plot of split_clicom()
- split_hdbscan(min_cluster_size: PositiveInt, min_samples: PositiveInt | None = 1, metric: str | Literal['Euclidean', 'Cosine', 'Pearson', 'Spearman', 'Manhattan', 'L1', 'L2', 'Jackknife', 'YS1', 'YR1', 'Sharpened_Cosine', 'Hamming'] = 'Euclidean', cluster_selection_epsilon: float = 0, cluster_selection_method: Literal['eom', 'leaf'] = 'eom', power_transform: bool = True, plot_style: Literal['all', 'std_area', 'std_bar'] = 'all', split_plots: bool = False, return_probabilities: bool = False, parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) Tuple[CountFilter, ...] | List[Tuple[CountFilter, ...] | ndarray] | None
Clusters the features in the CountFilter object using the HDBSCAN clustering algorithm, and then splits those features into multiple non-overlapping CountFilter objects, based on the clustering result.
- Parameters:
min_cluster_size (int) – the minimum size of clusters the algorithm will seek. Larger values will lead to fewer, larger clusters.
min_samples (int or None (default=1)) – the number of samples in a neighbourhood for a point to be considered a core point. Higher values will lead to a more conservative clustering result, with more points being classified as noise. If min_samples is None, the algorithm will pick a value automatically
metric (str (default='Euclidean')) – the distance metric used to determine similarity between data points. For a full list of supported distance metrics see the user guide.
cluster_selection_epsilon (float (default=0.0)) – a distance threshold below which clusters will be merged.
cluster_selection_method ('eom' or 'leaf' (default='eom')) – The method used to select clusters from the condensed tree. ‘eom’ will use an Excess of Mass algorithm to find the most persistent clusters. ‘leaf’ will select the leaves of the tree, providing the most fine-grained and homogenous clusters.
power_transform (bool (default=True)) – if True, RNAlysis will apply a power transform (Box-Cox) to the data prior to clustering.
plot_style ('all', 'std_area', or 'std_bar' (default='all')) – determines the visual style of the cluster expression plot.
split_plots (bool (default=False)) – if True, each discovered cluster will be plotted on its own. Otherwise, all clusters will be plotted in the same Figure.
return_probabilities (bool (default False)) – if True, the algorithm will return an array containing the probability with which each sample is a member of its assigned cluster, in addition to returning the clustering results. Points which were categorized as noise have probability 0.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Returns:
if return_probabilities is False, returns a tuple of CountFilter objects, each corresponding to a discovered cluster. Otherswise, returns a tuple of CountFilter objects, and a numpy array containing the probability values.
- Examples:
>>> from rnalysis import filtering >>> dev_stages = filtering.CountFilter('tests/test_files/elegans_developmental_stages.tsv') >>> dev_stages.filter_low_reads(100) Filtered 44072 features, leaving 2326 of the original 46398 features. Filtered inplace. >>> clusters = dev_stages.split_hdbscan(min_cluster_size=75,metric='yr1',power_transform=True) Found 14 clusters of average size 141.57. Number of unclustered genes is 344, which are 14.79% of the genes. Filtered 2019 features, leaving 307 of the original 2326 features. Filtering result saved to new object. Filtered 2122 features, leaving 204 of the original 2326 features. Filtering result saved to new object. Filtered 2146 features, leaving 180 of the original 2326 features. Filtering result saved to new object. Filtered 2168 features, leaving 158 of the original 2326 features. Filtering result saved to new object. Filtered 2173 features, leaving 153 of the original 2326 features. Filtering result saved to new object. Filtered 2176 features, leaving 150 of the original 2326 features. Filtering result saved to new object. Filtered 2183 features, leaving 143 of the original 2326 features. Filtering result saved to new object. Filtered 2192 features, leaving 134 of the original 2326 features. Filtering result saved to new object. Filtered 2200 features, leaving 126 of the original 2326 features. Filtering result saved to new object. Filtered 2234 features, leaving 92 of the original 2326 features. Filtering result saved to new object. Filtered 2238 features, leaving 88 of the original 2326 features. Filtering result saved to new object. Filtered 2241 features, leaving 85 of the original 2326 features. Filtering result saved to new object. Filtered 2244 features, leaving 82 of the original 2326 features. Filtering result saved to new object. Filtered 2246 features, leaving 80 of the original 2326 features. Filtering result saved to new object.
Example plot of split_hdbscan(plot_style=’all’)
Example plot of split_hdbscan()
- split_hierarchical(n_clusters: PositiveInt | List[PositiveInt] | Literal['gap', 'silhouette', 'calinski_harabasz', 'davies_bouldin', 'bic', 'distance'], metric: Literal['Euclidean', 'Cosine', 'Pearson', 'Spearman', 'Manhattan', 'L1', 'L2', 'Jackknife', 'YS1', 'YR1', 'Sharpened_Cosine'] = 'Euclidean', linkage: Literal['Single', 'Average', 'Complete', 'Ward'] = 'Average', power_transform: bool = True, distance_threshold: float | None = None, plot_style: Literal['all', 'std_area', 'std_bar'] = 'all', split_plots: bool = False, max_n_clusters_estimate: PositiveInt | Literal['auto'] = 'auto', parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) Tuple[CountFilter, ...] | Tuple[Tuple[CountFilter, ...], ...]
Clusters the features in the CountFilter object using the Hierarchical clustering algorithm, and then splits those features into multiple non-overlapping CountFilter objects, based on the clustering result.
- Parameters:
n_clusters (int, list of ints, 'distance', 'gap', 'silhouette', 'calinski_harabasz', 'davies_bouldin', or 'bic') – The number of clusters the algorithm will seek. If set to ‘distance’, the algorithm will derive the number of clusters from the distance threshold (see ‘distance_threshold’).
metric ('Euclidean', 'l1', 'l2', 'manhattan', or 'cosine', (default='Euclidean')) – the distance metric used to determine similarity between data points. If linkage is ‘ward’, only the ‘Euclidean’ metric is accepted. For a full list of supported distance metrics see the user guide.
linkage ('single', 'Average', 'complete', or 'ward' (default='Average')) – Which linkage criterion to use. The linkage criterion determines which distance to use between sets of observation. The algorithm will merge the pairs of cluster that minimize this criterion.
power_transform (bool (default=True)) – if True, RNAlysis will apply a power transform (Box-Cox) to the data prior to clustering.
distance_threshold (float or None (default=None)) – a distance threshold above which clusters will not be merged. If a number is specified, n_clusters must be None.
plot_style ('all', 'std_area', or 'std_bar' (default='all')) – determines the visual style of the cluster expression plot.
split_plots (bool (default=False)) – if True, each discovered cluster will be plotted on its own. Otherwise, all clusters will be plotted in the same Figure.
max_n_clusters_estimate (int or 'auto' (default='auto')) – the maximum number of clusters to test if trying to automatically estimate the optimal number of clusters. If `max_n_clusters_estimate`=’default’, an appropriate value will be picked automatically.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Returns:
if n_clusters is an int, returns a tuple of n_clusters CountFilter objects, each corresponding to a discovered cluster. If n_clusters is a list, returns one tuple of CountFilter objects per value in n_clusters.
- Examples:
>>> from rnalysis import filtering >>> dev_stages = filtering.CountFilter('tests/test_files/elegans_developmental_stages.tsv') >>> dev_stages.filter_low_reads(100) Filtered 44072 features, leaving 2326 of the original 46398 features. Filtered inplace. >>> clusters = dev_stages.split_hierarchical(n_clusters=13, metric='Euclidean',linkage='ward' ... ,power_transform=True) Filtered 1718 features, leaving 608 of the original 2326 features. Filtering result saved to new object. Filtered 1979 features, leaving 347 of the original 2326 features. Filtering result saved to new object. Filtered 2094 features, leaving 232 of the original 2326 features. Filtering result saved to new object. Filtered 2110 features, leaving 216 of the original 2326 features. Filtering result saved to new object. Filtered 2156 features, leaving 170 of the original 2326 features. Filtering result saved to new object. Filtered 2191 features, leaving 135 of the original 2326 features. Filtering result saved to new object. Filtered 2195 features, leaving 131 of the original 2326 features. Filtering result saved to new object. Filtered 2223 features, leaving 103 of the original 2326 features. Filtering result saved to new object. Filtered 2224 features, leaving 102 of the original 2326 features. Filtering result saved to new object. Filtered 2238 features, leaving 88 of the original 2326 features. Filtering result saved to new object. Filtered 2246 features, leaving 80 of the original 2326 features. Filtering result saved to new object. Filtered 2252 features, leaving 74 of the original 2326 features. Filtering result saved to new object. Filtered 2286 features, leaving 40 of the original 2326 features. Filtering result saved to new object.
Example plot of split_hierarchical(plot_style=’all’)
- align:
center
Example plot of split_hierarchical()
- split_kmeans(n_clusters: PositiveInt | List[PositiveInt] | Literal['gap', 'silhouette', 'calinski_harabasz', 'davies_bouldin', 'bic'], n_init: PositiveInt = 3, max_iter: PositiveInt = 300, random_seed: NonNegativeInt | None = None, power_transform: bool = True, plot_style: Literal['all', 'std_area', 'std_bar'] = 'all', split_plots: bool = False, max_n_clusters_estimate: PositiveInt | Literal['auto'] = 'auto', parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) Tuple[CountFilter, ...] | Tuple[Tuple[CountFilter, ...], ...]
Clusters the features in the CountFilter object using the K-means clustering algorithm, and then splits those features into multiple non-overlapping CountFilter objects, based on the clustering result.
- Parameters:
n_clusters (int, list of ints, 'gap', 'silhouette', 'calinski_harabasz', 'davies_bouldin', or 'bic') – The number of clusters the algorithm will seek.
random_seed (Union[int, None] or None (default=None)) – determines random number generation for centroid initialization. Use an int to make the randomness deterministic.
n_init (int (default=3)) – number of time the k-medoids algorithm will be run with different medoid seeds. The final results will be the best output of n_init consecutive runs in terms of inertia.
max_iter (int (default=300)) – maximum number of iterations of the k-medoids algorithm for a single run.
power_transform (bool (default=True)) – if True, RNAlysis will apply a power transform (Box-Cox) to the data prior to clustering.
plot_style ('all', 'std_area', or 'std_bar' (default='all')) – determines the visual style of the cluster expression plot.
split_plots (bool (default=False)) – if True, each discovered cluster will be plotted on its own. Otherwise, all clusters will be plotted in the same Figure.
max_n_clusters_estimate (int or 'auto' (default='auto')) – the maximum number of clusters to test if trying to automatically estimate the optimal number of clusters. If `max_n_clusters_estimate`=’default’, an appropriate value will be picked automatically.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Returns:
if n_clusters is an int, returns a tuple of n_clusters CountFilter objects, each corresponding to a discovered cluster. If n_clusters is a list, returns one tuple of CountFilter objects per value in n_clusters.
- Examples:
>>> from rnalysis import filtering >>> dev_stages = filtering.CountFilter('tests/test_files/elegans_developmental_stages.tsv') >>> dev_stages.filter_low_reads(100) Filtered 44072 features, leaving 2326 of the original 46398 features. Filtered inplace. >>> clusters = dev_stages.split_kmeans(14,power_transform=True) Filtered 44072 features, leaving 2326 of the original 46398 features. Filtered inplace. Filtered 1801 features, leaving 525 of the original 2326 features. Filtering result saved to new object. Filtered 2010 features, leaving 316 of the original 2326 features. Filtering result saved to new object. Filtered 2059 features, leaving 267 of the original 2326 features. Filtering result saved to new object. Filtered 2102 features, leaving 224 of the original 2326 features. Filtering result saved to new object. Filtered 2185 features, leaving 141 of the original 2326 features. Filtering result saved to new object. Filtered 2186 features, leaving 140 of the original 2326 features. Filtering result saved to new object. Filtered 2200 features, leaving 126 of the original 2326 features. Filtering result saved to new object. Filtered 2219 features, leaving 107 of the original 2326 features. Filtering result saved to new object. Filtered 2225 features, leaving 101 of the original 2326 features. Filtering result saved to new object. Filtered 2225 features, leaving 101 of the original 2326 features. Filtering result saved to new object. Filtered 2241 features, leaving 85 of the original 2326 features. Filtering result saved to new object. Filtered 2250 features, leaving 76 of the original 2326 features. Filtering result saved to new object. Filtered 2259 features, leaving 67 of the original 2326 features. Filtering result saved to new object. Filtered 2276 features, leaving 50 of the original 2326 features. Filtering result saved to new object.
Example plot of split_kmeans(plot_style=’all’)
Example plot of split_kmeans()
- split_kmedoids(n_clusters: PositiveInt | List[PositiveInt] | Literal['gap', 'silhouette', 'calinski_harabasz', 'davies_bouldin', 'bic'], n_init: PositiveInt = 3, max_iter: PositiveInt = 300, random_seed: NonNegativeInt | None = None, metric: str | Literal['Euclidean', 'Cosine', 'Pearson', 'Spearman', 'Manhattan', 'L1', 'L2', 'Jackknife', 'YS1', 'YR1', 'Sharpened_Cosine', 'Hamming'] = 'Euclidean', power_transform: bool = True, plot_style: Literal['all', 'std_area', 'std_bar'] = 'all', split_plots: bool = False, max_n_clusters_estimate: PositiveInt | Literal['auto'] = 'auto', parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) Tuple[CountFilter, ...] | Tuple[Tuple[CountFilter, ...], ...]
Clusters the features in the CountFilter object using the K-medoids clustering algorithm, and then splits those features into multiple non-overlapping CountFilter objects, based on the clustering result.
- Parameters:
n_clusters (int, list of ints, 'gap', 'silhouette', 'calinski_harabasz', 'davies_bouldin', or 'bic') – The number of clusters the algorithm will seek.
random_seed (Union[int, None] or None (default=None)) – determines random number generation for centroid initialization. Use an int to make the randomness deterministic.
n_init (int (default=3)) – number of time the k-medoids algorithm will be run with different medoid seeds. The final results will be the best output of n_init consecutive runs in terms of inertia.
max_iter (int (default=300)) – maximum number of iterations of the k-medoids algorithm for a single run.
metric (str (default='Euclidean')) – the distance metric used to determine similarity between data points. For a full list of supported distance metrics see the user guide.
power_transform (bool (default=True)) – if True, RNAlysis will apply a power transform (Box-Cox) to the data prior to clustering.
plot_style ('all', 'std_area', or 'std_bar' (default='all')) – determines the visual style of the cluster expression plot.
split_plots (bool (default=False)) – if True, each discovered cluster will be plotted on its own. Otherwise, all clusters will be plotted in the same Figure.
max_n_clusters_estimate (int or 'auto' (default='auto')) – the maximum number of clusters to test if trying to automatically estimate the optimal number of clusters. If `max_n_clusters_estimate`=’default’, an appropriate value will be picked automatically.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Returns:
if n_clusters is an int, returns a tuple of n_clusters CountFilter objects, each corresponding to a discovered cluster. If n_clusters is a list, returns one tuple of CountFilter objects per value in n_clusters.
- Examples:
>>> from rnalysis import filtering >>> dev_stages = filtering.CountFilter('tests/test_files/elegans_developmental_stages.tsv') >>> dev_stages.filter_low_reads(100) Filtered 44072 features, leaving 2326 of the original 46398 features. Filtered inplace. >>> clusters = dev_stages.split_kmedoids(n_clusters=14, metric='spearman', power_transform=True) Filtered 1967 features, leaving 359 of the original 2326 features. Filtering result saved to new object. Filtered 2020 features, leaving 306 of the original 2326 features. Filtering result saved to new object. Filtered 2071 features, leaving 255 of the original 2326 features. Filtering result saved to new object. Filtered 2131 features, leaving 195 of the original 2326 features. Filtering result saved to new object. Filtered 2145 features, leaving 181 of the original 2326 features. Filtering result saved to new object. Filtered 2157 features, leaving 169 of the original 2326 features. Filtering result saved to new object. Filtered 2159 features, leaving 167 of the original 2326 features. Filtering result saved to new object. Filtered 2182 features, leaving 144 of the original 2326 features. Filtering result saved to new object. Filtered 2190 features, leaving 136 of the original 2326 features. Filtering result saved to new object. Filtered 2192 features, leaving 134 of the original 2326 features. Filtering result saved to new object. Filtered 2229 features, leaving 97 of the original 2326 features. Filtering result saved to new object. Filtered 2252 features, leaving 74 of the original 2326 features. Filtering result saved to new object. Filtered 2268 features, leaving 58 of the original 2326 features. Filtering result saved to new object. Filtered 2275 features, leaving 51 of the original 2326 features. Filtering result saved to new object.
Example plot of split_kmedoids(plot_style=’all’)
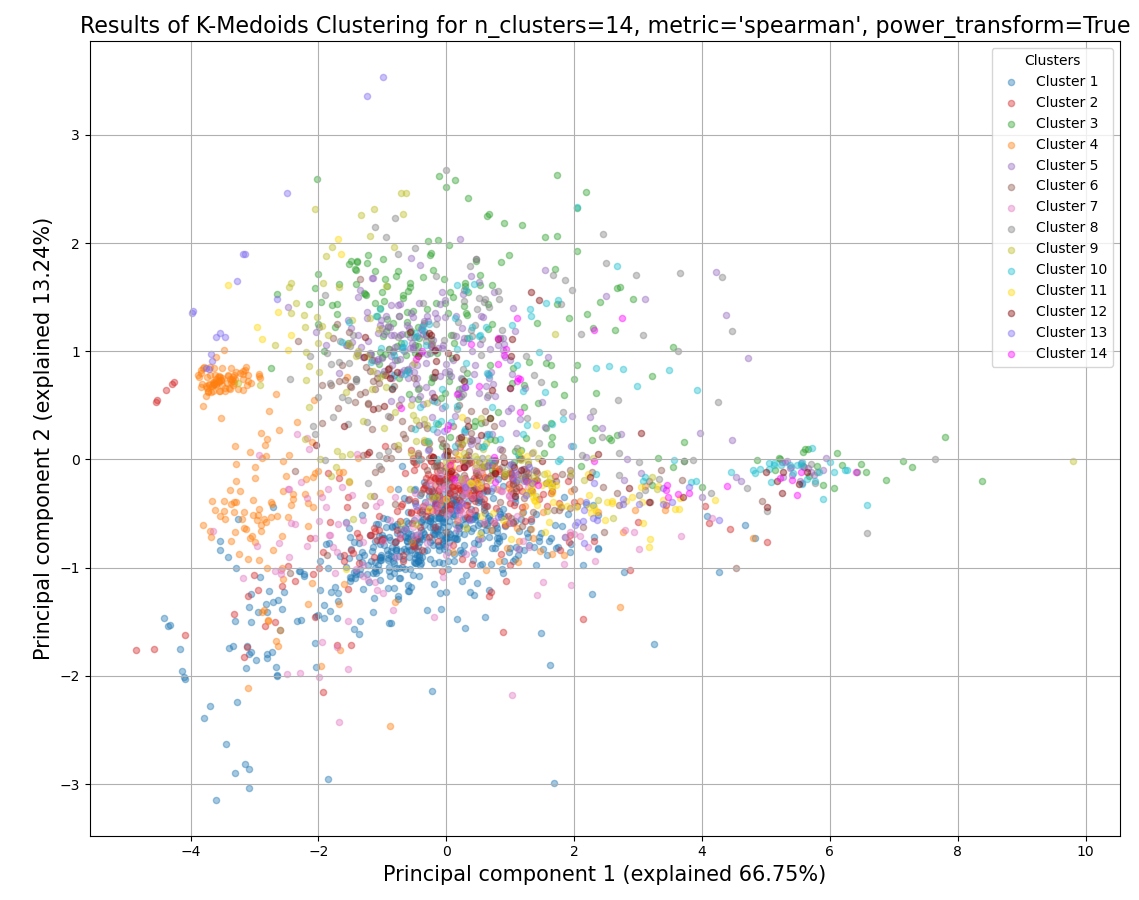
Example plot of split_kmedoids()
- symmetric_difference(other: Filter | set, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the WBGene indices that exist either in the first Filter object/set OR the second, but NOT in both (set symmetric difference).
- Parameters:
other (Filter or set.) – a second Filter object/set to calculate symmetric difference with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the features that exist in exactly one Filter object. If ‘str’, returns a string of the features that exist in exactly one Filter object, delimited by a comma.
- Return type:
set or str
- Returns:
a set/string of the features that that exist t in exactly one Filter. (set symmetric difference).
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter("tests/test_files/test_deseq.csv") >>> counts = filtering.CountFilter('tests/test_files/counted.csv') >>> # calculate difference and return a set >>> d.symmetric_difference(counts) {'WBGene00000017', 'WBGene00077504', 'WBGene00000024', 'WBGene00000010', 'WBGene00000020', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00007078', 'WBGene00000029', 'WBGene00000006', 'WBGene00007064', 'WBGene00000019', 'WBGene00000004', 'WBGene00007066', 'WBGene00014997', 'WBGene00000023', 'WBGene00007074', 'WBGene00000025', 'WBGene00043989', 'WBGene00043988', 'WBGene00000014', 'WBGene00000027', 'WBGene00000021', 'WBGene00044022', 'WBGene00007079', 'WBGene00000012', 'WBGene00000005', 'WBGene00077503', 'WBGene00000026', 'WBGene00000003', 'WBGene00000002', 'WBGene00077502', 'WBGene00044951', 'WBGene00007077', 'WBGene00000007', 'WBGene00000008', 'WBGene00007076', 'WBGene00000013', 'WBGene00043990', 'WBGene00043987', 'WBGene00007071', 'WBGene00000011', 'WBGene00000015', 'WBGene00000018', 'WBGene00000016', 'WBGene00000028', 'WBGene00007075', 'WBGene00000022', 'WBGene00000009'}
- tail(n: PositiveInt = 5) DataFrame
Return the last n rows of the Filter object. See polars.DataFrame.tail documentation.
- Parameters:
n (positive int, default 5) – Number of rows to show.
- Return type:
pl.DataFrame
- Returns:
returns the last n rows of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> d.tail() shape: (5, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬────────────┬────────────┬────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪════════════╪════════════╪════════════╡ │ WBGene00000025 ┆ 2236.185837 ┆ 2.477374 ┆ 0.129606 ┆ 19.114626 ┆ 1.9100e-81 ┆ 1.4600e-78 │ │ WBGene00000026 ┆ 343.648987 ┆ -4.037191 ┆ 0.219781 ┆ -18.369115 ┆ 2.3200e-75 ┆ 1.7000e-72 │ │ WBGene00000027 ┆ 175.142856 ┆ 6.352044 ┆ 0.347777 ┆ 18.264706 ┆ 1.5800e-74 ┆ 1.1200e-71 │ │ WBGene00000028 ┆ 219.1632 ┆ 3.913657 ┆ 0.217802 ┆ 17.968851 ┆ 3.4200e-72 ┆ 2.3200e-69 │ │ WBGene00000029 ┆ 1066.242402 ┆ -2.811281 ┆ 0.158284 ┆ -17.761002 ┆ 1.4200e-70 ┆ 9.2900e-68 │ └────────────────┴─────────────┴────────────────┴──────────┴────────────┴────────────┴────────────┘
>>> d.tail(8) # returns the last 8 rows shape: (8, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬────────────┬────────────┬────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪════════════╪════════════╪════════════╡ │ WBGene00000022 ┆ 365.813048 ┆ 6.101303 ┆ 0.291484 ┆ 20.931891 ┆ 2.7400e-97 ┆ 2.4000e-94 │ │ WBGene00000023 ┆ 3168.566714 ┆ 3.906719 ┆ 0.190439 ┆ 20.514331 ┆ 1.6000e-93 ┆ 1.3400e-90 │ │ WBGene00000024 ┆ 221.925724 ┆ 4.801676 ┆ 0.246313 ┆ 19.494189 ┆ 1.2300e-84 ┆ 9.8200e-82 │ │ WBGene00000025 ┆ 2236.185837 ┆ 2.477374 ┆ 0.129606 ┆ 19.114626 ┆ 1.9100e-81 ┆ 1.4600e-78 │ │ WBGene00000026 ┆ 343.648987 ┆ -4.037191 ┆ 0.219781 ┆ -18.369115 ┆ 2.3200e-75 ┆ 1.7000e-72 │ │ WBGene00000027 ┆ 175.142856 ┆ 6.352044 ┆ 0.347777 ┆ 18.264706 ┆ 1.5800e-74 ┆ 1.1200e-71 │ │ WBGene00000028 ┆ 219.1632 ┆ 3.913657 ┆ 0.217802 ┆ 17.968851 ┆ 3.4200e-72 ┆ 2.3200e-69 │ │ WBGene00000029 ┆ 1066.242402 ┆ -2.811281 ┆ 0.158284 ┆ -17.761002 ┆ 1.4200e-70 ┆ 9.2900e-68 │ └────────────────┴─────────────┴────────────────┴──────────┴────────────┴────────────┴────────────┘
- text_filters(column: ColumnName, operator: Literal['equals', 'contains', 'starts with', 'ends with'], value: str, opposite: bool = False, inplace: bool = True)
Applay a text filter (equals, contains, starts with, ends with) on a particular column in the Filter object.
- Parameters:
column (str) – name of the column to filter by
operator (str: 'eq' / 'equals' / '=', 'ct' / 'contains' / 'in', 'sw' / 'starts with', 'ew' / 'ends with') – the operator to filter the column by (equals, contains, starts with, ends with)
value (number (int or float)) – the value to filter by
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/text_filters.csv') >>> # keep only rows that have a value that starts with 'AC3' in the column 'name'. >>> filt.text_filters('name','sw','AC3') Filtered 17 features, leaving 5 of the original 22 features. Filtered inplace.
- transform(function: Literal['Box-Cox', 'log2', 'log10', 'ln', 'Standardize'] | Callable, columns: ColumnNames | Literal['all'] = 'all', inplace: bool = True, **function_kwargs)
Transform the values in the Filter object with the specified function.
- Parameters:
function (Callable or str ('logx' for base-x log of the data + 1, 'box-cox' for Box-Cox transform of the data + 1, 'standardize' for standardization)) – The function or function name to be applied.
columns (str, list of str, or 'all' (default='all')) – The columns to which the transform should be applied.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
function_kwargs – Any additional keyworded arguments taken by the supplied function.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/counted.csv') >>> filt_log10 = filt.transform('log10', inplace=False) Transformed 22 features. Transformation result saved to new object. >>> filt.transform(lambda x: x+1, columns=['cond1','cond4']) Transformed 22 features. Transformed inplace.
- translate_gene_ids(translate_to: str | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'], translate_from: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', remove_unmapped_genes: bool = False, inplace: bool = True)
Translates gene names/IDs from one type to another. Mapping is done using the UniProtKB Gene ID Mapping service. You can choose to optionally drop from the table all rows that failed to be translated.
- Parameters:
translate_to (str) – the gene ID type to translate gene names/IDs to. For example: UniProtKB, Ensembl, Wormbase.
translate_from (str or 'auto' (default='auto')) – the gene ID type to translate gene names/IDs from. For example: UniProtKB, Ensembl, Wormbase. If translate_from=’auto’, RNAlysis will attempt to automatically determine the gene ID type of the features in the table.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be translated will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and translated instance of the Filter object.
- property triplicates
Returns a nested list of the column names in the CountFilter, grouped by alphabetical order into triplicates. For example, if counts.columns is [‘A_rep1’,’A_rep2’,’A_rep3’,’B_rep1’,’B_rep2’,_B_rep3’], then counts.triplicates will be [[‘A_rep1’,’A_rep2’,’A_rep3’],[‘B_rep1’,’B_rep2’,_B_rep3’]]
- union(*others: Filter | set, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the union of features between multiple Filter objects/sets (the features that exist in at least one of the Filter objects/sets).
- Parameters:
others (Filter or set objects.) – Objects to calculate union with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the union features. If ‘str’, returns a string of the union WBGene indices, delimited by a comma.
- Return type:
set or str
- Returns:
a set/string of the WBGene indices that exist in at least one of the Filter objects.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # calculate union and return a set >>> d.union(counts) {'WBGene00000017', 'WBGene00000021', 'WBGene00044022', 'WBGene00077504', 'WBGene00000012', 'WBGene00000024', 'WBGene00007079', 'WBGene00000010', 'WBGene00000020', 'WBGene00000005', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00077503', 'WBGene00007078', 'WBGene00000026', 'WBGene00000029', 'WBGene00000002', 'WBGene00000003', 'WBGene00000006', 'WBGene00007064', 'WBGene00077502', 'WBGene00044951', 'WBGene00000007', 'WBGene00000008', 'WBGene00000019', 'WBGene00007077', 'WBGene00000004', 'WBGene00007066', 'WBGene00007076', 'WBGene00000013', 'WBGene00014997', 'WBGene00000023', 'WBGene00043990', 'WBGene00007074', 'WBGene00000025', 'WBGene00000011', 'WBGene00043987', 'WBGene00007071', 'WBGene00000015', 'WBGene00000018', 'WBGene00043989', 'WBGene00043988', 'WBGene00000014', 'WBGene00000016', 'WBGene00000027', 'WBGene00000028', 'WBGene00007075', 'WBGene00000022', 'WBGene00000009'}
- violin_plot(samples: GroupedColumns | Literal['all'] = 'all', ylabel: str = '$\\log_10$(normalized reads + 1)', title: str | Literal['auto'] = 'auto', title_fontsize: float = 20, label_fontsize: float = 16, tick_fontsize: float = 12) Figure
Generates a violin plot of the specified samples in the CountFilter object in log10 scale. Can plot both single samples and average multiple replicates. It is recommended to use this function on normalized values and not on absolute read values. Box inside the violin plot indicates 25% and 75% percentiles, and the white dot indicates the median.
- Parameters:
samples ('all' or list.) – A list of the sample names and/or grouped sample names to be plotted. All specified samples must be present in the CountFilter object. To average multiple replicates of the same condition, they can be grouped in an inner list. Example input: [[‘SAMPLE1A’, ‘SAMPLE1B’, ‘SAMPLE1C’], [‘SAMPLE2A’, ‘SAMPLE2B’, ‘SAMPLE2C’],’SAMPLE3’ , ‘SAMPLE6’]
ylabel (str (default='log10(normalized reads + 1)')) – the label of the Y axis.
title (str or 'auto' (default='auto')) – The title of the plot. If ‘auto’, a title will be generated automatically.
title_fontsize (float (default=30)) – determines the font size of the graph title.
label_fontsize (float (default=15) :param tick_fontsize: determines the font size of the X and Y tick labels.) – determines the font size of the X and Y axis labels.
- Return type:
a matplotlib Figure.
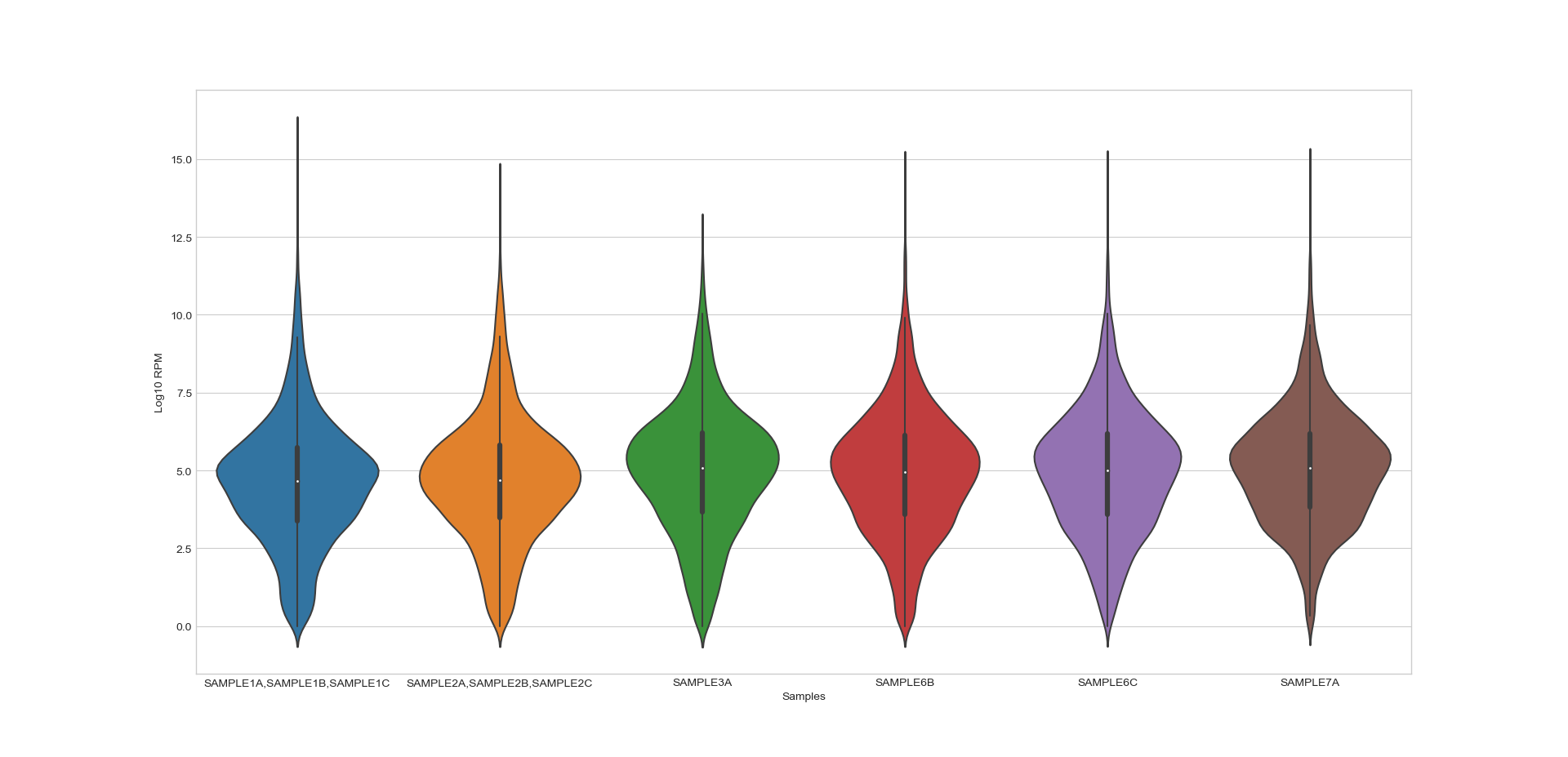
Example plot of violin_plot()
- class rnalysis.filtering.DESeqFilter(fname: str | Path | tuple, drop_columns: str | List[str] = None, log2fc_col: str | Literal['log2FoldChange', 'logFC'] = 'log2FoldChange', padj_col: str | Literal['padj', 'adj.P.Val'] = 'padj', pval_col: str | Literal['pvalue', 'P.Value'] = 'pvalue', suppress_warnings: bool = False)
Bases:
FilterA class that receives a DESeq output file and can filter it according to various characteristics.
Attributes
- df: polars DataFrame
A DataFrame that contains the DESeq output file contents. The DataFrame is modified upon usage of filter operations.
- shape: tuple (rows, columns)
The dimensions of df.
- columns: list
The columns of df.
- fname: pathlib.Path
The path and filename for the purpose of saving df as a csv file. Updates automatically when filter operations are applied.
- index_set: set
All of the indices in the current DataFrame (which were not removed by previously used filter methods) as a set.
- index_string: string
A string of all feature indices in the current DataFrame separated by newline.
- static _from_string(msg: str = '', delimiter: str = '\n')
Takes a manual string input from the user, and then splits it using a delimiter into a list of values.
- param msg:
a promprt to be printed to the user
- param delimiter:
the delimiter used to separate the values. Default is ‘
- ‘
- return:
A list of the comma-seperated values the user inserted.
- _inplace(new_df: DataFrame, opposite: bool, inplace: bool, suffix: str, printout_operation: str = 'filter', **filter_update_kwargs)
Executes the user’s choice whether to filter in-place or create a new instance of the Filter object.
- Parameters:
new_df (pl.DataFrame) – the post-filtering DataFrame
opposite (bool) – Determines whether to return the filtration ,or its opposite.
inplace (bool) – Determines whether to filter in-place or not.
suffix (str) – The suffix to be added to the filename
- Returns:
If inplace is False, returns a new instance of the Filter object.
- _set_ops(others, return_type: Literal['set', 'str'], op: Any, **kwargs)
Apply the supplied set operation (union/intersection/difference/symmetric difference) to the supplied objects.
- Parameters:
others (Filter or set objects.) – the other objects to apply the set operation to
return_type ('set' or 'str') – the return type of the output
op (function (set.union, set.intersection, set.difference or set.symmetric_difference)) – the set operation
kwargs – any additional keyworded arguments to be supplied to the set operation.
- Returns:
a set/string of indices resulting from the set operation
- Return type:
set or str
- _sort(by: str | List[str], ascending: bool | List[bool] = True, na_position: str = 'last')
Sort the rows by the values of specified column or columns.
- Parameters:
by (str or list of str) – Names of the column or columns to sort by.
ascending (bool or list of bool, default True) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
inplace (bool, default True) – If True, perform operation in-place. Otherwise, returns a sorted copy of the Filter object without modifying the original.
- Returns:
None if inplace=True, a sorted Filter object otherwise.
- annotate_from_gtf(gtf_path: str | Path, attribute: Literal['gene_biotype', 'transcript_biotype', 'biotype', 'gene_name', 'gene_id', 'transcript_id', 'chromosome', 'source', 'strand'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', column_name: str | None = None, inplace: bool = True)
Adds a new column to the table, annotating each feature with the value of a GTF/GFF attribute (for example: the biotype, chromosome, strand, or source of each gene). Features that are not found in the annotation file are annotated with a missing value.
- Parameters:
gtf_path (str or Path) – Path to your GTF/GFF annotation file. The file should match the type of gene names/IDs you use in your table.
attribute (str (default='gene_biotype')) – name of the attribute to annotate with. Standard column-9 attributes (such as ‘gene_biotype’, ‘gene_name’, or any custom key in your file) are supported, as well as the reserved names ‘chromosome’, ‘source’ and ‘strand’, which are read from the fixed columns of the annotation file.
feature_type ('gene' or 'transcript' (default='gene')) – determines whether the features/rows in your data table describe individual genes or transcripts.
column_name (str or None (default=None)) – the name of the new annotation column. If not specified, the attribute name is used. If a column with this name already exists in the table, it will be overwritten.
inplace (bool (default=True)) – If True (default), the annotation column will be added to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and annotated instance of the Filter object.
- biotypes_from_gtf(gtf_path: str | Path, attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', long_format: bool = False) DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- biotypes_from_ref_table(long_format: bool = False, ref: str | Path | Literal['predefined'] = 'predefined') DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :param ref: Name of the biotype reference table used to determine biotype. Default is ce11 (included in the package). :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # short-form view >>> d.biotypes_from_ref_table(ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 2) ┌────────────────┬───────┐ │ biotype ┆ count │ │ --- ┆ --- │ │ str ┆ u32 │ ╞════════════════╪═══════╡ │ unknown ┆ 1 │ │ protein_coding ┆ 26 │ │ pseudogene ┆ 1 │ └────────────────┴───────┘
>>> # long-form view >>> d.biotypes_from_ref_table(long_format=True,ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 49) ┌───────────┬───────────┬───────────┬───────────┬───┬───────────┬───────────┬───────────┬──────────┐ │ biotype ┆ baseMean_ ┆ baseMean_ ┆ baseMean_ ┆ … ┆ padj_25% ┆ padj_50% ┆ padj_75% ┆ padj_max │ │ --- ┆ count ┆ mean ┆ std ┆ ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ --- ┆ --- ┆ --- ┆ ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ │ ┆ f64 ┆ f64 ┆ f64 ┆ ┆ ┆ ┆ ┆ │ ╞═══════════╪═══════════╪═══════════╪═══════════╪═══╪═══════════╪═══════════╪═══════════╪══════════╡ │ protein_c ┆ 26.0 ┆ 1823.0896 ┆ 1796.5207 ┆ … ┆ 3.8506e-1 ┆ 1.1350e-1 ┆ 1.0051e-9 ┆ 9.2900e- │ │ oding ┆ ┆ 09 ┆ 8 ┆ ┆ 76 ┆ 35 ┆ 0 ┆ 68 │ │ pseudogen ┆ 1.0 ┆ 2688.0437 ┆ null ┆ … ┆ 1.8000e-9 ┆ 1.8000e-9 ┆ 1.8000e-9 ┆ 1.8000e- │ │ e ┆ ┆ 01 ┆ ┆ ┆ 4 ┆ 4 ┆ 4 ┆ 94 │ │ unknown ┆ 1.0 ┆ 2085.9950 ┆ null ┆ … ┆ 3.0700e-1 ┆ 3.0700e-1 ┆ 3.0700e-1 ┆ 3.0700e- │ │ ┆ ┆ 94 ┆ ┆ ┆ 52 ┆ 52 ┆ 52 ┆ 152 │ └───────────┴───────────┴───────────┴───────────┴───┴───────────┴───────────┴───────────┴──────────┘
- property columns: list
The columns of df.
- Returns:
a list of the columns in the Filter object.
- Return type:
list
- describe(percentiles: float | List[float] = (0.01, 0.25, 0.5, 0.75, 0.99)) DataFrame
Generate descriptive statistics that summarize the central tendency, dispersion and shape of the dataset’s distribution, excluding NaN values. For more information see the documentation of polars.DataFrame.describe.
- Parameters:
percentiles (list-like of floats (default=(0.01, 0.25, 0.5, 0.75, 0.99))) – The percentiles to include in the output. All should fall between 0 and 1. The default is [.25, .5, .75], which returns the 25th, 50th, and 75th percentiles.
- Returns:
Summary statistics of the dataset.
- Return type:
pl.DataFrame
- Examples:
>>> from rnalysis import filtering >>> import numpy as np >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.describe() cond1 cond2 cond3 cond4 count 22.000000 22.000000 22.000000 22.000000 mean 2515.590909 2209.227273 4230.227273 3099.818182 std 4820.512674 4134.948493 7635.832664 5520.394522 min 0.000000 0.000000 0.000000 0.000000 1% 0.000000 0.000000 0.000000 0.000000 25% 6.000000 6.250000 1.250000 0.250000 50% 57.500000 52.500000 23.500000 21.000000 75% 2637.000000 2479.000000 6030.500000 4669.750000 99% 15054.950000 12714.290000 21955.390000 15603.510000 max 15056.000000 12746.000000 22027.000000 15639.000000
>>> # show the deciles (10%, 20%, 30%... 90%) of the columns >>> counts.describe(percentiles=np.arange(0.1, 1, 0.1)) cond1 cond2 cond3 cond4 count 22.000000 22.000000 22.000000 22.000000 mean 2515.590909 2209.227273 4230.227273 3099.818182 std 4820.512674 4134.948493 7635.832664 5520.394522 min 0.000000 0.000000 0.000000 0.000000 10% 0.000000 0.200000 0.000000 0.000000 20% 1.400000 3.200000 1.000000 0.000000 30% 15.000000 15.700000 2.600000 1.000000 40% 28.400000 26.800000 14.000000 9.000000 50% 57.500000 52.500000 23.500000 21.000000 60% 82.000000 106.800000 44.000000 33.000000 70% 484.200000 395.500000 305.000000 302.500000 80% 3398.600000 3172.600000 7981.400000 6213.000000 90% 8722.100000 7941.800000 16449.500000 12129.900000 max 15056.000000 12746.000000 22027.000000 15639.000000
- df
- difference(*others: Filter | set, return_type: Literal['set', 'str'] = 'set', inplace: bool = False)
Keep only the features that exist in the first Filter object/set but NOT in the others. Can be done inplace on the first Filter object, or return a set/string of features.
- Parameters:
others (Filter or set objects.) – Objects to calculate difference with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the features that exist only in the first Filter object. If ‘str’, returns a string of the WBGene indices that exist only in the first Filter object, delimited by a comma.
inplace (bool, default False) – If True, filtering will be applied to the current Filter object. If False (default), the function will return a set/str that contains the intersecting features.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that exist only in the first Filter object/set (set difference).
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter("tests/test_files/test_deseq.csv") >>> counts = filtering.CountFilter('tests/test_files/counted.csv') >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> # calculate difference and return a set >>> d.difference(counts, a_set) {'WBGene00007063', 'WBGene00007064', 'WBGene00007066', 'WBGene00007067', 'WBGene00007069', 'WBGene00007071', 'WBGene00007074', 'WBGene00007075', 'WBGene00007076', 'WBGene00007077', 'WBGene00007078', 'WBGene00007079', 'WBGene00014997', 'WBGene00043987', 'WBGene00043988', 'WBGene00043989', 'WBGene00043990', 'WBGene00044022', 'WBGene00044951', 'WBGene00077502', 'WBGene00077503', 'WBGene00077504'}
# calculate difference and filter in-place >>> d.difference(counts, a_set, inplace=True) Filtered 2 features, leaving 26 of the original 28 features. Filtered inplace.
- drop_columns(columns: ColumnNames, inplace: bool = True)
Drop specific columns from the table.
- Parameters:
columns (str or list of str) – The names of the column/columns to be dropped fro mthe table.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_abs_log2_fold_change(abslog2fc: float = 1, opposite: bool = False, inplace: bool = True)
Filters out all features whose absolute log2 fold change is below the indicated threshold. For example: if log2fc is 2.0, all features whose log2 fold change is between 1 and -1 (went up less than two-fold or went down less than two-fold) will be filtered out.
- Parameters:
abslog2fc – The threshold absolute log2 fold change for filtering out a feature. Float or int. All features whose absolute log2 fold change is lower than log2fc will be filtered out.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current DESeqFilter object. If False, the function will return a new DESeqFilter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of DESeqFilter.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter('tests/test_files/sample_deseq.csv') >>> d.filter_abs_log2_fold_change(2) # keep only rows whose log2(fold change) is >=2 or <=-2 Filtered 1 features, leaving 28 of the original 29 features. Filtered inplace.
- filter_biotype_from_gtf(gtf_path: str | Path, biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
biotype (str or list of strings) – the biotypes which will not be filtered out.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- filter_biotype_from_ref_table(biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
- Parameters:
biotype (string or list of strings) – the biotypes which will not be filtered out.
ref – Name of the biotype reference file used to determine biotypes. Default is the path defined by the user in the settings.yaml file.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows whose biotype is 'protein_coding' >>> counts.filter_biotype_from_ref_table('protein_coding',ref='tests/test_files/biotype_ref_table_for_tests.csv') Filtered 9 features, leaving 13 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows whose biotype is 'protein_coding' or 'pseudogene' >>> counts.filter_biotype_from_ref_table(['protein_coding','pseudogene'],ref='tests/test_files/biotype_ref_table_for_tests.csv') Filtered 0 features, leaving 22 of the original 22 features. Filtered inplace.
- filter_by_attribute(attributes: str | List[str] = None, mode: Literal['union', 'intersection'] = 'union', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters features according to user-defined attributes from an Attribute Reference Table. When multiple attributes are given, filtering can be done in ‘union’ mode (where features that belong to at least one attribute are not filtered out), or in ‘intersection’ mode (where only features that belong to ALL attributes are not filtered out). To learn more about user-defined attributes and Attribute Reference Tables, read the user guide.
- Parameters:
attributes (string or list of strings, which are column titles in the user-defined Attribute Reference Table.) – attributes to filter by.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
ref (str or pathlib.Path (default='predefined')) – filename/path of the attribute reference table to be used as reference.
opposite (bool (default=False)) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to the attribute 'attribute1' >>> counts.filter_by_attribute('attribute1',ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 15 features, leaving 7 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to the attributes 'attribute1' OR 'attribute3' (union) >>> counts.filter_by_attribute(['attribute1','attribute3'],ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 14 features, leaving 8 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to both attributes 'attribute1' AND 'attribute3' (intersection) >>> counts.filter_by_attribute(['attribute1','attribute3'],mode='intersection', ... ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 19 features, leaving 3 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that DON'T belong to either 'attribute1','attribute3' or both >>> counts.filter_by_attribute(['attribute1','attribute3'],ref='tests/test_files/attr_ref_table_for_examples.csv', ... opposite=True) Filtered 8 features, leaving 14 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that DON'T belong to both 'attribute1' AND 'attribute3' >>> counts.filter_by_attribute(['attribute1','attribute3'],mode='intersection', ... ref='tests/test_files/attr_ref_table_for_examples.csv',opposite=True) Filtered 3 features, leaving 19 of the original 22 features. Filtered inplace.
- filter_by_go_annotations(go_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', propagate_annotations: bool = True, evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', opposite: bool = False, inplace: bool = True)
Filters genes according to GO annotations, keeping only genes that are annotated with a specific GO term. When multiple GO terms are given, filtering can be done in ‘union’ mode (where genes that belong to at least one GO term are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL GO terms are not filtered out).
- Parameters:
go_ids (str or list of str)
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :param propagate_annotations: determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/ :type propagate_annotations: ‘classic’, ‘elim’, ‘weight’, ‘all.m’, or ‘no’ (default=’elim’) :param evidence_types: only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or ‘any’ (default=’any’) :param excluded_evidence_types: annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type excluded_evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or None (default=None) :param databases: only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current FeatureSet object. If False, the function will return a new FeatureSet instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the FeatureSet object.
- filter_by_gtf_attribute(gtf_path: str | Path, attribute: Literal['gene_biotype', 'transcript_biotype', 'biotype', 'gene_name', 'gene_id', 'transcript_id', 'chromosome', 'source', 'strand'] | str = 'gene_biotype', value: str | List[str] = 'protein_coding', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters the features in the table by any attribute described in a GTF/GFF annotation file, keeping only features whose attribute matches one of the specified values (for example: keep only genes on a specific chromosome or strand, from a specific source, or of a specific biotype). This is a generalization of filter_biotype_from_gtf to any GTF/GFF attribute.
- Parameters:
gtf_path (str or Path) – Path to your GTF/GFF annotation file. The file should match the type of gene names/IDs you use in your table.
attribute (str (default='gene_biotype')) – name of the attribute to filter by. Standard column-9 attributes (such as ‘gene_biotype’, ‘gene_name’, or any custom key in your file) are supported, as well as the reserved names ‘chromosome’, ‘source’ and ‘strand’, which are read from the fixed columns of the annotation file.
value (str or list of strings) – the attribute value/values which will NOT be filtered out. For example, to keep only the features on chromosomes ‘chr1’ and ‘chr2’, set attribute=’chromosome’ and value=[‘chr1’, ‘chr2’].
feature_type ('gene' or 'transcript' (default='gene')) – determines whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- filter_by_kegg_annotations(kegg_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', opposite: bool = False, inplace: bool = True)
Filters genes according to KEGG pathways, keeping only genes that belong to specific KEGG pathway. When multiple KEGG IDs are given, filtering can be done in ‘union’ mode (where genes that belong to at least one pathway are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL pathways are not filtered out).
- Parameters:
kegg_ids (str or list of str) – the KEGG pathway IDs according to which the table will be filtered. An example for a legal KEGG pathway ID would be ‘path:cel04020’ for the C. elegans calcium signaling pathway.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the Filter object.
- filter_by_row_name(row_names: str | List[str], opposite: bool = False, inplace: bool = True)
Filter out specific rows from the table by their name (index).
- Parameters:
row_names (str or list of str) – list of row names to be removed from the table.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_duplicate_ids(keep: Literal['first', 'last', 'neither'] = 'first', opposite: bool = False, inplace: bool = True)
Filter out rows with duplicate names/IDs (index).
- Parameters:
keep ('first', 'last', or 'neither' (default='first')) – determines which of the duplicates to keep for each group of duplicates. ‘first’ will keep the first duplicate found for each group; ‘last’ will keep the last; and ‘neither’ will remove all of the values in the group.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_fold_change_direction(direction: Literal['pos', 'neg'] = 'pos', opposite: bool = False, inplace: bool = True)
Filters out features according to the direction in which they changed between the two conditions.
- Parameters:
direction – ‘pos’ or ‘neg’. If ‘pos’, will keep only features that have positive log2foldchange. If ‘neg’, will keep only features that have negative log2foldchange.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current DESeqFilter object. If False, the function will return a new DESeqFilter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of DESeqFilter.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter('tests/test_files/sample_deseq.csv') >>> d.filter_fold_change_direction('pos') # keep only rows with a positive log2(fold change) value Filtered 3 features, leaving 26 of the original 29 features. Filtered inplace.
>>> d = filtering.DESeqFilter('tests/test_files/sample_deseq.csv') >>> d.filter_fold_change_direction('neg') # keep only rows with a negative log2(fold change) value Filtered 27 features, leaving 2 of the original 29 features. Filtered inplace.
>>> d = filtering.DESeqFilter('tests/test_files/sample_deseq.csv') >>> d.filter_fold_change_direction('pos', opposite=True) # keep only rows with a non-positive log2(fold change) value Filtered 26 features, leaving 3 of the original 29 features. Filtered inplace.
- filter_missing_values(columns: ColumnNames | Literal['all'] = 'all', opposite: bool = False, inplace: bool = True)
Remove all rows whose values in the specified columns are missing (NaN).
:param columns:name/names of the columns to check for missing values. :type opposite: bool :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/test_deseq_with_nan.csv') >>> filt_no_nan = filt.filter_missing_values(inplace=False) Filtered 3 features, leaving 25 of the original 28 features. Filtering result saved to new object. >>> filt_no_nan_basemean = filt.filter_missing_values(columns='baseMean', inplace=False) Filtered 1 features, leaving 27 of the original 28 features. Filtering result saved to new object. >>> filt_no_nan_basemean_pval = filt.filter_missing_values(columns=['baseMean','pvalue'], inplace=False) Filtered 2 features, leaving 26 of the original 28 features. Filtering result saved to new object.
- filter_percentile(percentile: Fraction, column: ColumnName, interpolate: 'nearest', 'higher', 'lower', 'midpoint', 'linear' = 'linear', opposite: bool = False, inplace: bool = True)
Removes all entries above the specified percentile in the specified column. For example, if the column were ‘pvalue’ and the percentile was 0.5, then all features whose pvalue is above the median pvalue will be filtered out.
- Parameters:
percentile (float between 0 and 1) – The percentile that all features above it will be filtered out.
column (str) – Name of the DataFrame column according to which the filtering will be performed.
interpolate ('nearest', 'higher', 'lower', 'midpoint' or 'linear' (default='linear')) – interpolation method to use when the desired quantile lies between two data points.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # keep only the rows whose value in the column 'log2FoldChange' is below the 75th percentile >>> d.filter_percentile(0.75,'log2FoldChange') Filtered 7 features, leaving 21 of the original 28 features. Filtered inplace.
>>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # keep only the rows vulse value in the column 'log2FoldChange' is above the 25th percentile >>> d.filter_percentile(0.25,'log2FoldChange',opposite=True) Filtered 7 features, leaving 21 of the original 28 features. Filtered inplace.
- filter_significant(alpha: Fraction = 0.1, opposite: bool = False, inplace: bool = True)
Removes all features which did not change significantly, according to the provided alpha.
- Parameters:
alpha – the significance threshold to determine which genes will be filtered. between 0 and 1.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current DESeqFilter object. If False, the function will return a new DESeqFilter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of DESeqFilter.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter('tests/test_files/sample_deseq.csv') >>> d.filter_significant(0.1) # keep only rows whose adjusted p-value is <=0.1 Filtered 4 features, leaving 25 of the original 29 features. Filtered inplace.
>>> d = filtering.DESeqFilter('tests/test_files/sample_deseq.csv')
>>> d.filter_significant(0.1, opposite=True) # keep only rows whose adjusted p-value is >0.1 Filtered 25 features, leaving 4 of the original 29 features. Filtered inplace.
- filter_top_n(by: ColumnNames, n: PositiveInt = 100, ascending: bool | List[bool] = True, na_position: str = 'last', opposite: bool = False, inplace: bool = True)
Sort the rows by the values of specified column or columns, then keep only the top ‘n’ rows.
- Parameters:
by (name of column/columns (str/List[str])) – Names of the column or columns to sort and then filter by.
n (int) – How many features to keep in the Filter object.
ascending (bool or list of bools (default=True)) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of Filter.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only the 10 rows with the highest values in the columns 'cond1' >>> counts.filter_top_n(by='cond1',n=10, ascending=False) Filtered 12 features, leaving 10 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only the 10 rows which have the lowest values in the columns 'cond1' >>> # and then the highest values in the column 'cond2' >>> counts.filter_top_n(by=['cond1','cond2'],n=10, ascending=[True,False]) Filtered 12 features, leaving 10 of the original 22 features. Filtered inplace.
- find_paralogs_ensembl(organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True)
Find paralogs within the same species using the Ensembl database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
- Returns:
DataFrame describing all discovered paralog mappings.
- find_paralogs_panther(organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto')
Find paralogs within the same species using the PantherDB database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- Returns:
DataFrame describing all discovered paralog mappings.
- fname
- head(n: PositiveInt = 5) DataFrame
Return the first n rows of the Filter object. See polars.DataFrame.head documentation.
- Parameters:
n (positive int, default 5) – Number of rows to show.
- Returns:
returns the first n rows of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> d.head() shape: (5, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬───────────┬─────────────┬─────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪═══════════╪═════════════╪═════════════╡ │ WBGene00000002 ┆ 6820.755327 ┆ 7.567762 ┆ 0.142257 ┆ 53.197692 ┆ 0.0 ┆ 0.0 │ │ WBGene00000003 ┆ 3049.62567 ┆ 9.138071 ┆ 0.245989 ┆ 37.148359 ┆ 4.6600e-302 ┆ 4.2800e-298 │ │ WBGene00000004 ┆ 1432.911791 ┆ 8.111737 ┆ 0.246801 ┆ 32.867521 ┆ 6.4000e-237 ┆ 3.9200e-233 │ │ WBGene00000005 ┆ 4028.154186 ┆ 6.534112 ┆ 0.202467 ┆ 32.272543 ┆ 1.7000e-228 ┆ 7.8000e-225 │ │ WBGene00000006 ┆ 1230.58524 ┆ 7.157428 ┆ 0.227948 ┆ 31.399325 ┆ 2.0700e-216 ┆ 7.5900e-213 │ └────────────────┴─────────────┴────────────────┴──────────┴───────────┴─────────────┴─────────────┘
>>> d.head(3) # return only the first 3 rows shape: (3, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬───────────┬─────────────┬─────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪═══════════╪═════════════╪═════════════╡ │ WBGene00000002 ┆ 6820.755327 ┆ 7.567762 ┆ 0.142257 ┆ 53.197692 ┆ 0.0 ┆ 0.0 │ │ WBGene00000003 ┆ 3049.62567 ┆ 9.138071 ┆ 0.245989 ┆ 37.148359 ┆ 4.6600e-302 ┆ 4.2800e-298 │ │ WBGene00000004 ┆ 1432.911791 ┆ 8.111737 ┆ 0.246801 ┆ 32.867521 ┆ 6.4000e-237 ┆ 3.9200e-233 │ └────────────────┴─────────────┴────────────────┴──────────┴───────────┴─────────────┴─────────────┘
- property index_set: set
Returns all of the features in the current DataFrame (which were not removed by previously used filter methods) as a set. if any duplicate features exist in the filter object (same WBGene appears more than once), the corresponding WBGene index will appear in the returned set ONLY ONCE.
- Returns:
A set of WBGene names.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> myset = counts.index_set >>> print(myset) {'WBGene00044022', 'WBGene00077504', 'WBGene00007079', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00077503', 'WBGene00007078', 'WBGene00007064', 'WBGene00077502', 'WBGene00044951', 'WBGene00007077', 'WBGene00007066', 'WBGene00007076', 'WBGene00014997', 'WBGene00043990', 'WBGene00007074', 'WBGene00043987', 'WBGene00007071', 'WBGene00043989', 'WBGene00043988', 'WBGene00007075'}
- property index_string: str
Returns a string of all feature indices in the current DataFrame, sorted by their current order in the FIlter object, and separated by newline.
- This includes all of the feature indices which were not filtered out by previously-used filter methods.
if any duplicate features exist in the filter object (same index appears more than once),
the corresponding index will appear in the returned string ONLY ONCE.
- Returns:
A string of WBGene indices separated by newlines (\n).
For example, “WBGene00000001\nWBGene00000003\nWBGene12345678”.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.sort(by='cond1',ascending=False) >>> mystring = counts.index_string >>> print(mystring) WBGene00007075 WBGene00043988 WBGene00043990 WBGene00007079 WBGene00007076 WBGene00043989 WBGene00007063 WBGene00007077 WBGene00007078 WBGene00007071 WBGene00007064 WBGene00007066 WBGene00007074 WBGene00043987 WBGene00007067 WBGene00014997 WBGene00044022 WBGene00077503 WBGene00077504 WBGene00077502 WBGene00007069 WBGene00044951
- intersection(*others: Filter | set, return_type: Literal['set', 'str'] = 'set', inplace: bool = False)
Keep only the features that exist in ALL of the given Filter objects/sets. Can be done either inplace on the first Filter object, or return a set/string of features.
- Parameters:
others (Filter or set objects.) – Objects to calculate intersection with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the intersecting features. If ‘str’, returns a string of the intersecting features, delimited by a comma.
inplace (bool (default=False)) – If True, the function will be applied in-place to the current Filter object. If False (default), the function will return a set/str that contains the intersecting indices.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that intersect between the given Filter objects/sets.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> # calculate intersection and return a set >>> d.intersection(a_set) {'WBGene00000002', 'WBGene00000003'}
# calculate intersection and filter in-place >>> d.intersection(a_set, inplace=True) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
- log2fc_col
name of the log2 fold change column
- majority_vote_intersection(*others: Filter | set, majority_threshold: float = 0.5, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the features that appear in at least (majority_threhold * 100)% of the given Filter objects/sets. Majority-vote intersection with majority_threshold=0 is equivalent to Union. Majority-vote intersection with majority_threshold=1 is equivalent to Intersection.
- Parameters:
others (Filter or set objects.) – Objects to calculate intersection with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the intersecting WBGene indices. If ‘str’, returns a string of the intersecting indices, delimited by a comma.
majority_threshold (float (default=0.5)) – The threshold that determines what counts as majority. Features will be returned only if they appear in at least (majority_threshold * 100)% of the given Filter objects/sets.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that uphold majority vote intersection between two given Filter objects/sets.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> b_set = {'WBGene00000002','WBGene00000004'} >>> # calculate majority-vote intersection and return a set >>> d.majority_vote_intersection(a_set, b_set, majority_threshold=2/3) {'WBGene00000002', 'WBGene00000003', 'WBGene00000004'}
- map_orthologs_ensembl(map_to_organism: str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'], map_from_organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the Ensembl database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_orthoinspector(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the OrthoInspector database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_panther(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_least_diverged: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PantherDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_least_diverged (bool (default=True)) – if True (default), RNAlysis will only fetch ortholog mappings that were flagged as a ‘least diverged ortholog’ on the PantherDB database. You can read more about this flag on the PantherDB website: https://www.pantherdb.org/genes/
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_phylomedb(map_to_organism: str | int | Literal, map_from_organism: Literal['auto'] | str | int | Literal = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', consistency_score_threshold: Fraction = 0.5, filter_consistency_score: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PhylomeDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- param map_to_organism:
organism name or NCBI taxon ID of the target species for ortholog mapping.
- type map_to_organism:
str or int
- param map_from_organism:
organism name or NCBI taxon ID of the input genes’ source species.
- type map_from_organism:
str or int
- param gene_id_type:
the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- type gene_id_type:
str or ‘auto’ (default=’auto’)
- ` :param consistency_score_threshold: the minimum consistency score required for an ortholog mapping to be considered valid. Consistency scores are calculated by PhylomeDB and represent the confidence of the ortholog mapping. setting consistency_score_threshold to 0 will keep all mappings. You can read more about PhylomeDB consistency score on the PhylomeDB website: orthology.phylomedb.org/help
- type consistency_score_threshold:
float between 0 and 1 (default=0.5)
- param filter_consistency_score:
if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest consistency score.
- type filter_consistency_score:
bool (default=True)
- param non_unique_mode:
How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
- type non_unique_mode:
‘first’, ‘last’, ‘random’, or ‘none’ (default=’first’)
- param remove_unmapped_genes:
if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
- type remove_unmapped_genes:
bool (default=False)
- type inplace:
bool (default=True)
- param inplace:
If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- return:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- number_filters(column: ColumnName, operator: Literal['greater than', 'equals', 'lesser than', 'abs greater than'], value: float, opposite: bool = False, inplace: bool = True)
Applay a number filter (greater than, equal, lesser than) on a particular column in the Filter object.
- Parameters:
column (str) – name of the column to filter by
operator (str: 'gt' / 'greater than' / '>', 'eq' / 'equals' / '=', 'lt' / 'lesser than' / '<') – the operator to filter the column by (greater than, equal or lesser than)
value (float) – the value to filter by
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','gt',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
>>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','greater than',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
>>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','>',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
- padj_col
name of the adjusted p-value column
- print_features()
Print the feature indices in the Filter object, sorted by their current order in the FIlter object, and separated by newline.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.sort(by='cond1',ascending=False) >>> counts.print_features() WBGene00007075 WBGene00043988 WBGene00043990 WBGene00007079 WBGene00007076 WBGene00043989 WBGene00007063 WBGene00007077 WBGene00007078 WBGene00007071 WBGene00007064 WBGene00007066 WBGene00007074 WBGene00043987 WBGene00007067 WBGene00014997 WBGene00044022 WBGene00077503 WBGene00077504 WBGene00077502 WBGene00007069 WBGene00044951
- pval_col
name of the p-value column
- pval_histogram(adjusted_pvals: bool = False, bin_size: Fraction = 0.05, title: str | Literal['auto'] = 'auto') Figure
Plots a histogram of the p-values in the DESeqFilter object. This is often used to troubleshoot the results of a differential expression analysis. For more information about interpreting p-value histograms, see the following blog post by / David Robinson: https://varianceexplained.org/statistics/interpreting-pvalue-histogram/
- Parameters:
adjusted_pvals (bool (default=False)) – if True, will plot a histogram of the adjusted p-values instead of the raw p-values.
bin_size (float between 0 and 1 (default=0.05)) – determines the size of the bins in the histogram.
title (str or 'auto' (default='auto')) – the title of the histogram. If ‘auto’, will be set automatically.
- save_csv(alt_filename: None | str | Path = None)
Saves the current filtered data to a .csv file.
- Parameters:
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- save_parquet(alt_filename: None | str | Path = None)
Saves the current filtered data to a .parquet file.
- Parameters:
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- save_table(suffix: Literal['.csv', '.tsv', '.parquet'] = '.csv', alt_filename: None | str | Path = None)
Save the current filtered data table.
- Parameters:
suffix ('.csv', '.tsv', or '.parquet' (default='.csv')) – the file suffix
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- sort(by: str | List[str], ascending: bool | List[bool] = True, na_position: str = 'last', inplace: bool = True)
Sort the rows by the values of specified column or columns.
- Parameters:
by (str or list of str) – Names of the column or columns to sort by.
ascending (bool or list of bool (default=True)) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
inplace (bool (default=True)) – If True, perform operation in-place. Otherwise, returns a sorted copy of the Filter object without modifying the original.
- Returns:
None if inplace=True, a sorted Filter object otherwise.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.head() cond1 cond2 cond3 cond4 WBGene00007063 633 451 365 388 WBGene00007064 60 57 20 23 WBGene00044951 0 0 0 1 WBGene00007066 55 266 46 39 WBGene00007067 15 13 1 0 >>> counts.sort(by='cond1',ascending=True) >>> counts.head() cond1 cond2 cond3 cond4 WBGene00044951 0 0 0 1 WBGene00077504 0 0 0 0 WBGene00007069 0 2 1 0 WBGene00077502 0 0 0 0 WBGene00077503 1 4 2 0
- split_by_attribute(attributes: str | List[str], ref: str | Path | Literal['predefined'] = 'predefined') tuple
Splits the features in the Filter object into multiple Filter objects, each corresponding to one of the specified Attribute Reference Table attributes. Each new Filter object will contain only features that belong to its Attribute Reference Table attribute.
- Parameters:
attributes (list of strings) – list of attribute names from the Attribute Reference Table to filter by.
ref – filename/path of the reference table to be used as reference.
- Return type:
Tuple[filtering.Filter]
- Returns:
A tuple of Filter objects, each containing only features that match one Attribute Reference Table attribute; the Filter objects are returned in the same order the attributes were given in.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> attribute1,attribute2 = counts.split_by_attribute(['attribute1','attribute2'], ... ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 15 features, leaving 7 of the original 22 features. Filtering result saved to new object. Filtered 20 features, leaving 2 of the original 22 features. Filtering result saved to new object.
- split_by_percentile(percentile: Fraction, column: ColumnName, interpolate: 'nearest', 'higher', 'lower', 'midpoint', 'linear' = 'linear') tuple
Splits the features in the Filter object into two non-overlapping Filter objects: one containing features below the specified percentile in the specfieid column, and the other containing features about the specified percentile in the specified column.
- Parameters:
percentile (float between 0 and 1) – The percentile that all features above it will be filtered out.
column (str) – Name of the DataFrame column according to which the filtering will be performed.
- Return type:
Tuple[filtering.Filter, filtering.Filter]
- Returns:
a tuple of two Filter objects: the first contains all of the features below the specified percentile, and the second contains all of the features above and equal to the specified percentile.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> below, above = d.split_by_percentile(0.75,'log2FoldChange') Filtered 7 features, leaving 21 of the original 28 features. Filtering result saved to new object. Filtered 21 features, leaving 7 of the original 28 features. Filtering result saved to new object.
- split_fold_change_direction() tuple
Splits the features in the DESeqFilter object into two non-overlapping DESeqFilter objects, based on the direction of their log2foldchange. The first object will contain only features with a positive log2foldchange, the second object will contain only features with a negative log2foldchange.
- Return type:
Tuple[filtering.DESeqFilter, filteirng.DESeqFilter]
- Returns:
a tuple containing two DESeqFilter objects: the first has only features with positive log2 fold change, and the other has only features with negative log2 fold change.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter('tests/test_files/test_deseq.csv') >>> pos, neg = d.split_fold_change_direction() Filtered 2 features, leaving 26 of the original 28 features. Filtering result saved to new object. Filtered 26 features, leaving 2 of the original 28 features. Filtering result saved to new object.
- symmetric_difference(other: Filter | set, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the WBGene indices that exist either in the first Filter object/set OR the second, but NOT in both (set symmetric difference).
- Parameters:
other (Filter or set.) – a second Filter object/set to calculate symmetric difference with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the features that exist in exactly one Filter object. If ‘str’, returns a string of the features that exist in exactly one Filter object, delimited by a comma.
- Return type:
set or str
- Returns:
a set/string of the features that that exist t in exactly one Filter. (set symmetric difference).
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter("tests/test_files/test_deseq.csv") >>> counts = filtering.CountFilter('tests/test_files/counted.csv') >>> # calculate difference and return a set >>> d.symmetric_difference(counts) {'WBGene00000017', 'WBGene00077504', 'WBGene00000024', 'WBGene00000010', 'WBGene00000020', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00007078', 'WBGene00000029', 'WBGene00000006', 'WBGene00007064', 'WBGene00000019', 'WBGene00000004', 'WBGene00007066', 'WBGene00014997', 'WBGene00000023', 'WBGene00007074', 'WBGene00000025', 'WBGene00043989', 'WBGene00043988', 'WBGene00000014', 'WBGene00000027', 'WBGene00000021', 'WBGene00044022', 'WBGene00007079', 'WBGene00000012', 'WBGene00000005', 'WBGene00077503', 'WBGene00000026', 'WBGene00000003', 'WBGene00000002', 'WBGene00077502', 'WBGene00044951', 'WBGene00007077', 'WBGene00000007', 'WBGene00000008', 'WBGene00007076', 'WBGene00000013', 'WBGene00043990', 'WBGene00043987', 'WBGene00007071', 'WBGene00000011', 'WBGene00000015', 'WBGene00000018', 'WBGene00000016', 'WBGene00000028', 'WBGene00007075', 'WBGene00000022', 'WBGene00000009'}
- tail(n: PositiveInt = 5) DataFrame
Return the last n rows of the Filter object. See polars.DataFrame.tail documentation.
- Parameters:
n (positive int, default 5) – Number of rows to show.
- Return type:
pl.DataFrame
- Returns:
returns the last n rows of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> d.tail() shape: (5, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬────────────┬────────────┬────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪════════════╪════════════╪════════════╡ │ WBGene00000025 ┆ 2236.185837 ┆ 2.477374 ┆ 0.129606 ┆ 19.114626 ┆ 1.9100e-81 ┆ 1.4600e-78 │ │ WBGene00000026 ┆ 343.648987 ┆ -4.037191 ┆ 0.219781 ┆ -18.369115 ┆ 2.3200e-75 ┆ 1.7000e-72 │ │ WBGene00000027 ┆ 175.142856 ┆ 6.352044 ┆ 0.347777 ┆ 18.264706 ┆ 1.5800e-74 ┆ 1.1200e-71 │ │ WBGene00000028 ┆ 219.1632 ┆ 3.913657 ┆ 0.217802 ┆ 17.968851 ┆ 3.4200e-72 ┆ 2.3200e-69 │ │ WBGene00000029 ┆ 1066.242402 ┆ -2.811281 ┆ 0.158284 ┆ -17.761002 ┆ 1.4200e-70 ┆ 9.2900e-68 │ └────────────────┴─────────────┴────────────────┴──────────┴────────────┴────────────┴────────────┘
>>> d.tail(8) # returns the last 8 rows shape: (8, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬────────────┬────────────┬────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪════════════╪════════════╪════════════╡ │ WBGene00000022 ┆ 365.813048 ┆ 6.101303 ┆ 0.291484 ┆ 20.931891 ┆ 2.7400e-97 ┆ 2.4000e-94 │ │ WBGene00000023 ┆ 3168.566714 ┆ 3.906719 ┆ 0.190439 ┆ 20.514331 ┆ 1.6000e-93 ┆ 1.3400e-90 │ │ WBGene00000024 ┆ 221.925724 ┆ 4.801676 ┆ 0.246313 ┆ 19.494189 ┆ 1.2300e-84 ┆ 9.8200e-82 │ │ WBGene00000025 ┆ 2236.185837 ┆ 2.477374 ┆ 0.129606 ┆ 19.114626 ┆ 1.9100e-81 ┆ 1.4600e-78 │ │ WBGene00000026 ┆ 343.648987 ┆ -4.037191 ┆ 0.219781 ┆ -18.369115 ┆ 2.3200e-75 ┆ 1.7000e-72 │ │ WBGene00000027 ┆ 175.142856 ┆ 6.352044 ┆ 0.347777 ┆ 18.264706 ┆ 1.5800e-74 ┆ 1.1200e-71 │ │ WBGene00000028 ┆ 219.1632 ┆ 3.913657 ┆ 0.217802 ┆ 17.968851 ┆ 3.4200e-72 ┆ 2.3200e-69 │ │ WBGene00000029 ┆ 1066.242402 ┆ -2.811281 ┆ 0.158284 ┆ -17.761002 ┆ 1.4200e-70 ┆ 9.2900e-68 │ └────────────────┴─────────────┴────────────────┴──────────┴────────────┴────────────┴────────────┘
- text_filters(column: ColumnName, operator: Literal['equals', 'contains', 'starts with', 'ends with'], value: str, opposite: bool = False, inplace: bool = True)
Applay a text filter (equals, contains, starts with, ends with) on a particular column in the Filter object.
- Parameters:
column (str) – name of the column to filter by
operator (str: 'eq' / 'equals' / '=', 'ct' / 'contains' / 'in', 'sw' / 'starts with', 'ew' / 'ends with') – the operator to filter the column by (equals, contains, starts with, ends with)
value (number (int or float)) – the value to filter by
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/text_filters.csv') >>> # keep only rows that have a value that starts with 'AC3' in the column 'name'. >>> filt.text_filters('name','sw','AC3') Filtered 17 features, leaving 5 of the original 22 features. Filtered inplace.
- transform(function: Literal['Box-Cox', 'log2', 'log10', 'ln', 'Standardize'] | Callable, columns: ColumnNames | Literal['all'] = 'all', inplace: bool = True, **function_kwargs)
Transform the values in the Filter object with the specified function.
- Parameters:
function (Callable or str ('logx' for base-x log of the data + 1, 'box-cox' for Box-Cox transform of the data + 1, 'standardize' for standardization)) – The function or function name to be applied.
columns (str, list of str, or 'all' (default='all')) – The columns to which the transform should be applied.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
function_kwargs – Any additional keyworded arguments taken by the supplied function.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/counted.csv') >>> filt_log10 = filt.transform('log10', inplace=False) Transformed 22 features. Transformation result saved to new object. >>> filt.transform(lambda x: x+1, columns=['cond1','cond4']) Transformed 22 features. Transformed inplace.
- translate_gene_ids(translate_to: str | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'], translate_from: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', remove_unmapped_genes: bool = False, inplace: bool = True)
Translates gene names/IDs from one type to another. Mapping is done using the UniProtKB Gene ID Mapping service. You can choose to optionally drop from the table all rows that failed to be translated.
- Parameters:
translate_to (str) – the gene ID type to translate gene names/IDs to. For example: UniProtKB, Ensembl, Wormbase.
translate_from (str or 'auto' (default='auto')) – the gene ID type to translate gene names/IDs from. For example: UniProtKB, Ensembl, Wormbase. If translate_from=’auto’, RNAlysis will attempt to automatically determine the gene ID type of the features in the table.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be translated will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and translated instance of the Filter object.
- union(*others: Filter | set, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the union of features between multiple Filter objects/sets (the features that exist in at least one of the Filter objects/sets).
- Parameters:
others (Filter or set objects.) – Objects to calculate union with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the union features. If ‘str’, returns a string of the union WBGene indices, delimited by a comma.
- Return type:
set or str
- Returns:
a set/string of the WBGene indices that exist in at least one of the Filter objects.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # calculate union and return a set >>> d.union(counts) {'WBGene00000017', 'WBGene00000021', 'WBGene00044022', 'WBGene00077504', 'WBGene00000012', 'WBGene00000024', 'WBGene00007079', 'WBGene00000010', 'WBGene00000020', 'WBGene00000005', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00077503', 'WBGene00007078', 'WBGene00000026', 'WBGene00000029', 'WBGene00000002', 'WBGene00000003', 'WBGene00000006', 'WBGene00007064', 'WBGene00077502', 'WBGene00044951', 'WBGene00000007', 'WBGene00000008', 'WBGene00000019', 'WBGene00007077', 'WBGene00000004', 'WBGene00007066', 'WBGene00007076', 'WBGene00000013', 'WBGene00014997', 'WBGene00000023', 'WBGene00043990', 'WBGene00007074', 'WBGene00000025', 'WBGene00000011', 'WBGene00043987', 'WBGene00007071', 'WBGene00000015', 'WBGene00000018', 'WBGene00043989', 'WBGene00043988', 'WBGene00000014', 'WBGene00000016', 'WBGene00000027', 'WBGene00000028', 'WBGene00007075', 'WBGene00000022', 'WBGene00000009'}
- volcano_plot(alpha: Fraction = 0.1, log2fc_threshold: float | None = 1, title: str | Literal['auto'] = 'auto', title_fontsize: float = 16, label_fontsize: float = 16, tick_fontsize: float = 12, annotation_fontsize: float = 10, point_size: float = 10, opacity: Fraction = 0.65, interactive: bool = True, show_cursor: bool = False) Figure
Plots a volcano plot (log2(fold change) vs -log10(adj. p-value)) of the DESeqFilter object. Significantly upregulated features are colored in red, and significantly downregulated features are colored in blue. If the plot is generated in interactive mode, data points can be labeled by clicking on them.
- Parameters:
alpha (float between 0 and 1) – the significance threshold to paint data points as significantly up/down-regulated.
log2fc_threshold (non-negative float or None (default=1)) – the absolute log2(fold change) threshold to paint data as significantly up/down-regulated. if log2fc_threshold is None, no threshold will be used.
title (str or 'auto' (default='auto')) – The title of the plot. If ‘auto’, a title will be generated automatically.
title_fontsize (float (default=30)) – determines the font size of the graph title.
label_fontsize (float (default=15) :param tick_fontsize: determines the font size of the X and Y tick labels.) – determines the font size of the X and Y axis labels.
annotation_fontsize (float (default=10)) – determines the font size of the point annotations created in interactive mode.
opacity (determines the opacity of the points in the scatter plot. 0 indicates completely transparent, while 1 indicates completely opaque.) – float between 0 and 1 (default=0.65)
point_size (float (default=10)) – determines the size of the points in the scatter plot
interactive (bool (default=True)) – if True, turns on interactive mode. While in interactive mode, you can click on a data point to label it with its gene name/ID, or click on a labeled data point to unlabel it.
show_cursor (bool (default=False)) – if True, show the cursor position on the plot during interactive mode
- Return type:
A matplotlib Figure
Example plot of volcano_plot()
- class rnalysis.filtering.Filter(fname: str | Path, drop_columns: str | List[str] = None, suppress_warnings: bool = False)
Bases:
objectAn all-purpose Filter object.
Attributes
- df: polars DataFrame
A DataFrame that contains the DESeq output file contents. The DataFrame is modified upon usage of filter operations.
- shape: tuple (rows, columns)
The dimensions of df.
- columns: list
The columns of df.
- fname: pathlib.Path
The path and filename for the purpose of saving df as a csv file. Updates automatically when filter operations are applied.
- index_set: set
All of the indices in the current DataFrame (which were not removed by previously used filter methods) as a set.
- index_string: string
A string of all feature indices in the current DataFrame separated by newline.
- static _from_string(msg: str = '', delimiter: str = '\n')
Takes a manual string input from the user, and then splits it using a delimiter into a list of values.
- param msg:
a promprt to be printed to the user
- param delimiter:
the delimiter used to separate the values. Default is ‘
- ‘
- return:
A list of the comma-seperated values the user inserted.
- _inplace(new_df: DataFrame, opposite: bool, inplace: bool, suffix: str, printout_operation: str = 'filter', **filter_update_kwargs)
Executes the user’s choice whether to filter in-place or create a new instance of the Filter object.
- Parameters:
new_df (pl.DataFrame) – the post-filtering DataFrame
opposite (bool) – Determines whether to return the filtration ,or its opposite.
inplace (bool) – Determines whether to filter in-place or not.
suffix (str) – The suffix to be added to the filename
- Returns:
If inplace is False, returns a new instance of the Filter object.
- _set_ops(others, return_type: Literal['set', 'str'], op: Any, **kwargs)
Apply the supplied set operation (union/intersection/difference/symmetric difference) to the supplied objects.
- Parameters:
others (Filter or set objects.) – the other objects to apply the set operation to
return_type ('set' or 'str') – the return type of the output
op (function (set.union, set.intersection, set.difference or set.symmetric_difference)) – the set operation
kwargs – any additional keyworded arguments to be supplied to the set operation.
- Returns:
a set/string of indices resulting from the set operation
- Return type:
set or str
- _sort(by: str | List[str], ascending: bool | List[bool] = True, na_position: str = 'last')
Sort the rows by the values of specified column or columns.
- Parameters:
by (str or list of str) – Names of the column or columns to sort by.
ascending (bool or list of bool, default True) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
inplace (bool, default True) – If True, perform operation in-place. Otherwise, returns a sorted copy of the Filter object without modifying the original.
- Returns:
None if inplace=True, a sorted Filter object otherwise.
- annotate_from_gtf(gtf_path: str | Path, attribute: Literal['gene_biotype', 'transcript_biotype', 'biotype', 'gene_name', 'gene_id', 'transcript_id', 'chromosome', 'source', 'strand'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', column_name: str | None = None, inplace: bool = True)
Adds a new column to the table, annotating each feature with the value of a GTF/GFF attribute (for example: the biotype, chromosome, strand, or source of each gene). Features that are not found in the annotation file are annotated with a missing value.
- Parameters:
gtf_path (str or Path) – Path to your GTF/GFF annotation file. The file should match the type of gene names/IDs you use in your table.
attribute (str (default='gene_biotype')) – name of the attribute to annotate with. Standard column-9 attributes (such as ‘gene_biotype’, ‘gene_name’, or any custom key in your file) are supported, as well as the reserved names ‘chromosome’, ‘source’ and ‘strand’, which are read from the fixed columns of the annotation file.
feature_type ('gene' or 'transcript' (default='gene')) – determines whether the features/rows in your data table describe individual genes or transcripts.
column_name (str or None (default=None)) – the name of the new annotation column. If not specified, the attribute name is used. If a column with this name already exists in the table, it will be overwritten.
inplace (bool (default=True)) – If True (default), the annotation column will be added to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and annotated instance of the Filter object.
- biotypes_from_gtf(gtf_path: str | Path, attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', long_format: bool = False) DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- biotypes_from_ref_table(long_format: bool = False, ref: str | Path | Literal['predefined'] = 'predefined') DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :param ref: Name of the biotype reference table used to determine biotype. Default is ce11 (included in the package). :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # short-form view >>> d.biotypes_from_ref_table(ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 2) ┌────────────────┬───────┐ │ biotype ┆ count │ │ --- ┆ --- │ │ str ┆ u32 │ ╞════════════════╪═══════╡ │ unknown ┆ 1 │ │ protein_coding ┆ 26 │ │ pseudogene ┆ 1 │ └────────────────┴───────┘
>>> # long-form view >>> d.biotypes_from_ref_table(long_format=True,ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 49) ┌───────────┬───────────┬───────────┬───────────┬───┬───────────┬───────────┬───────────┬──────────┐ │ biotype ┆ baseMean_ ┆ baseMean_ ┆ baseMean_ ┆ … ┆ padj_25% ┆ padj_50% ┆ padj_75% ┆ padj_max │ │ --- ┆ count ┆ mean ┆ std ┆ ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ --- ┆ --- ┆ --- ┆ ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ │ ┆ f64 ┆ f64 ┆ f64 ┆ ┆ ┆ ┆ ┆ │ ╞═══════════╪═══════════╪═══════════╪═══════════╪═══╪═══════════╪═══════════╪═══════════╪══════════╡ │ protein_c ┆ 26.0 ┆ 1823.0896 ┆ 1796.5207 ┆ … ┆ 3.8506e-1 ┆ 1.1350e-1 ┆ 1.0051e-9 ┆ 9.2900e- │ │ oding ┆ ┆ 09 ┆ 8 ┆ ┆ 76 ┆ 35 ┆ 0 ┆ 68 │ │ pseudogen ┆ 1.0 ┆ 2688.0437 ┆ null ┆ … ┆ 1.8000e-9 ┆ 1.8000e-9 ┆ 1.8000e-9 ┆ 1.8000e- │ │ e ┆ ┆ 01 ┆ ┆ ┆ 4 ┆ 4 ┆ 4 ┆ 94 │ │ unknown ┆ 1.0 ┆ 2085.9950 ┆ null ┆ … ┆ 3.0700e-1 ┆ 3.0700e-1 ┆ 3.0700e-1 ┆ 3.0700e- │ │ ┆ ┆ 94 ┆ ┆ ┆ 52 ┆ 52 ┆ 52 ┆ 152 │ └───────────┴───────────┴───────────┴───────────┴───┴───────────┴───────────┴───────────┴──────────┘
- property columns: list
The columns of df.
- Returns:
a list of the columns in the Filter object.
- Return type:
list
- describe(percentiles: float | List[float] = (0.01, 0.25, 0.5, 0.75, 0.99)) DataFrame
Generate descriptive statistics that summarize the central tendency, dispersion and shape of the dataset’s distribution, excluding NaN values. For more information see the documentation of polars.DataFrame.describe.
- Parameters:
percentiles (list-like of floats (default=(0.01, 0.25, 0.5, 0.75, 0.99))) – The percentiles to include in the output. All should fall between 0 and 1. The default is [.25, .5, .75], which returns the 25th, 50th, and 75th percentiles.
- Returns:
Summary statistics of the dataset.
- Return type:
pl.DataFrame
- Examples:
>>> from rnalysis import filtering >>> import numpy as np >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.describe() cond1 cond2 cond3 cond4 count 22.000000 22.000000 22.000000 22.000000 mean 2515.590909 2209.227273 4230.227273 3099.818182 std 4820.512674 4134.948493 7635.832664 5520.394522 min 0.000000 0.000000 0.000000 0.000000 1% 0.000000 0.000000 0.000000 0.000000 25% 6.000000 6.250000 1.250000 0.250000 50% 57.500000 52.500000 23.500000 21.000000 75% 2637.000000 2479.000000 6030.500000 4669.750000 99% 15054.950000 12714.290000 21955.390000 15603.510000 max 15056.000000 12746.000000 22027.000000 15639.000000
>>> # show the deciles (10%, 20%, 30%... 90%) of the columns >>> counts.describe(percentiles=np.arange(0.1, 1, 0.1)) cond1 cond2 cond3 cond4 count 22.000000 22.000000 22.000000 22.000000 mean 2515.590909 2209.227273 4230.227273 3099.818182 std 4820.512674 4134.948493 7635.832664 5520.394522 min 0.000000 0.000000 0.000000 0.000000 10% 0.000000 0.200000 0.000000 0.000000 20% 1.400000 3.200000 1.000000 0.000000 30% 15.000000 15.700000 2.600000 1.000000 40% 28.400000 26.800000 14.000000 9.000000 50% 57.500000 52.500000 23.500000 21.000000 60% 82.000000 106.800000 44.000000 33.000000 70% 484.200000 395.500000 305.000000 302.500000 80% 3398.600000 3172.600000 7981.400000 6213.000000 90% 8722.100000 7941.800000 16449.500000 12129.900000 max 15056.000000 12746.000000 22027.000000 15639.000000
- df
polars.DataFrame with the data
- difference(*others: Filter | set, return_type: Literal['set', 'str'] = 'set', inplace: bool = False)
Keep only the features that exist in the first Filter object/set but NOT in the others. Can be done inplace on the first Filter object, or return a set/string of features.
- Parameters:
others (Filter or set objects.) – Objects to calculate difference with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the features that exist only in the first Filter object. If ‘str’, returns a string of the WBGene indices that exist only in the first Filter object, delimited by a comma.
inplace (bool, default False) – If True, filtering will be applied to the current Filter object. If False (default), the function will return a set/str that contains the intersecting features.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that exist only in the first Filter object/set (set difference).
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter("tests/test_files/test_deseq.csv") >>> counts = filtering.CountFilter('tests/test_files/counted.csv') >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> # calculate difference and return a set >>> d.difference(counts, a_set) {'WBGene00007063', 'WBGene00007064', 'WBGene00007066', 'WBGene00007067', 'WBGene00007069', 'WBGene00007071', 'WBGene00007074', 'WBGene00007075', 'WBGene00007076', 'WBGene00007077', 'WBGene00007078', 'WBGene00007079', 'WBGene00014997', 'WBGene00043987', 'WBGene00043988', 'WBGene00043989', 'WBGene00043990', 'WBGene00044022', 'WBGene00044951', 'WBGene00077502', 'WBGene00077503', 'WBGene00077504'}
# calculate difference and filter in-place >>> d.difference(counts, a_set, inplace=True) Filtered 2 features, leaving 26 of the original 28 features. Filtered inplace.
- drop_columns(columns: ColumnNames, inplace: bool = True)
Drop specific columns from the table.
- Parameters:
columns (str or list of str) – The names of the column/columns to be dropped fro mthe table.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_biotype_from_gtf(gtf_path: str | Path, biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
biotype (str or list of strings) – the biotypes which will not be filtered out.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- filter_biotype_from_ref_table(biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
- Parameters:
biotype (string or list of strings) – the biotypes which will not be filtered out.
ref – Name of the biotype reference file used to determine biotypes. Default is the path defined by the user in the settings.yaml file.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows whose biotype is 'protein_coding' >>> counts.filter_biotype_from_ref_table('protein_coding',ref='tests/test_files/biotype_ref_table_for_tests.csv') Filtered 9 features, leaving 13 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows whose biotype is 'protein_coding' or 'pseudogene' >>> counts.filter_biotype_from_ref_table(['protein_coding','pseudogene'],ref='tests/test_files/biotype_ref_table_for_tests.csv') Filtered 0 features, leaving 22 of the original 22 features. Filtered inplace.
- filter_by_attribute(attributes: str | List[str] = None, mode: Literal['union', 'intersection'] = 'union', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters features according to user-defined attributes from an Attribute Reference Table. When multiple attributes are given, filtering can be done in ‘union’ mode (where features that belong to at least one attribute are not filtered out), or in ‘intersection’ mode (where only features that belong to ALL attributes are not filtered out). To learn more about user-defined attributes and Attribute Reference Tables, read the user guide.
- Parameters:
attributes (string or list of strings, which are column titles in the user-defined Attribute Reference Table.) – attributes to filter by.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
ref (str or pathlib.Path (default='predefined')) – filename/path of the attribute reference table to be used as reference.
opposite (bool (default=False)) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to the attribute 'attribute1' >>> counts.filter_by_attribute('attribute1',ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 15 features, leaving 7 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to the attributes 'attribute1' OR 'attribute3' (union) >>> counts.filter_by_attribute(['attribute1','attribute3'],ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 14 features, leaving 8 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to both attributes 'attribute1' AND 'attribute3' (intersection) >>> counts.filter_by_attribute(['attribute1','attribute3'],mode='intersection', ... ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 19 features, leaving 3 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that DON'T belong to either 'attribute1','attribute3' or both >>> counts.filter_by_attribute(['attribute1','attribute3'],ref='tests/test_files/attr_ref_table_for_examples.csv', ... opposite=True) Filtered 8 features, leaving 14 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that DON'T belong to both 'attribute1' AND 'attribute3' >>> counts.filter_by_attribute(['attribute1','attribute3'],mode='intersection', ... ref='tests/test_files/attr_ref_table_for_examples.csv',opposite=True) Filtered 3 features, leaving 19 of the original 22 features. Filtered inplace.
- filter_by_go_annotations(go_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', propagate_annotations: bool = True, evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', opposite: bool = False, inplace: bool = True)
Filters genes according to GO annotations, keeping only genes that are annotated with a specific GO term. When multiple GO terms are given, filtering can be done in ‘union’ mode (where genes that belong to at least one GO term are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL GO terms are not filtered out).
- Parameters:
go_ids (str or list of str)
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :param propagate_annotations: determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/ :type propagate_annotations: ‘classic’, ‘elim’, ‘weight’, ‘all.m’, or ‘no’ (default=’elim’) :param evidence_types: only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or ‘any’ (default=’any’) :param excluded_evidence_types: annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type excluded_evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or None (default=None) :param databases: only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current FeatureSet object. If False, the function will return a new FeatureSet instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the FeatureSet object.
- filter_by_gtf_attribute(gtf_path: str | Path, attribute: Literal['gene_biotype', 'transcript_biotype', 'biotype', 'gene_name', 'gene_id', 'transcript_id', 'chromosome', 'source', 'strand'] | str = 'gene_biotype', value: str | List[str] = 'protein_coding', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters the features in the table by any attribute described in a GTF/GFF annotation file, keeping only features whose attribute matches one of the specified values (for example: keep only genes on a specific chromosome or strand, from a specific source, or of a specific biotype). This is a generalization of filter_biotype_from_gtf to any GTF/GFF attribute.
- Parameters:
gtf_path (str or Path) – Path to your GTF/GFF annotation file. The file should match the type of gene names/IDs you use in your table.
attribute (str (default='gene_biotype')) – name of the attribute to filter by. Standard column-9 attributes (such as ‘gene_biotype’, ‘gene_name’, or any custom key in your file) are supported, as well as the reserved names ‘chromosome’, ‘source’ and ‘strand’, which are read from the fixed columns of the annotation file.
value (str or list of strings) – the attribute value/values which will NOT be filtered out. For example, to keep only the features on chromosomes ‘chr1’ and ‘chr2’, set attribute=’chromosome’ and value=[‘chr1’, ‘chr2’].
feature_type ('gene' or 'transcript' (default='gene')) – determines whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- filter_by_kegg_annotations(kegg_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', opposite: bool = False, inplace: bool = True)
Filters genes according to KEGG pathways, keeping only genes that belong to specific KEGG pathway. When multiple KEGG IDs are given, filtering can be done in ‘union’ mode (where genes that belong to at least one pathway are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL pathways are not filtered out).
- Parameters:
kegg_ids (str or list of str) – the KEGG pathway IDs according to which the table will be filtered. An example for a legal KEGG pathway ID would be ‘path:cel04020’ for the C. elegans calcium signaling pathway.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the Filter object.
- filter_by_row_name(row_names: str | List[str], opposite: bool = False, inplace: bool = True)
Filter out specific rows from the table by their name (index).
- Parameters:
row_names (str or list of str) – list of row names to be removed from the table.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_duplicate_ids(keep: Literal['first', 'last', 'neither'] = 'first', opposite: bool = False, inplace: bool = True)
Filter out rows with duplicate names/IDs (index).
- Parameters:
keep ('first', 'last', or 'neither' (default='first')) – determines which of the duplicates to keep for each group of duplicates. ‘first’ will keep the first duplicate found for each group; ‘last’ will keep the last; and ‘neither’ will remove all of the values in the group.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_missing_values(columns: ColumnNames | Literal['all'] = 'all', opposite: bool = False, inplace: bool = True)
Remove all rows whose values in the specified columns are missing (NaN).
:param columns:name/names of the columns to check for missing values. :type opposite: bool :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/test_deseq_with_nan.csv') >>> filt_no_nan = filt.filter_missing_values(inplace=False) Filtered 3 features, leaving 25 of the original 28 features. Filtering result saved to new object. >>> filt_no_nan_basemean = filt.filter_missing_values(columns='baseMean', inplace=False) Filtered 1 features, leaving 27 of the original 28 features. Filtering result saved to new object. >>> filt_no_nan_basemean_pval = filt.filter_missing_values(columns=['baseMean','pvalue'], inplace=False) Filtered 2 features, leaving 26 of the original 28 features. Filtering result saved to new object.
- filter_percentile(percentile: Fraction, column: ColumnName, interpolate: 'nearest', 'higher', 'lower', 'midpoint', 'linear' = 'linear', opposite: bool = False, inplace: bool = True)
Removes all entries above the specified percentile in the specified column. For example, if the column were ‘pvalue’ and the percentile was 0.5, then all features whose pvalue is above the median pvalue will be filtered out.
- Parameters:
percentile (float between 0 and 1) – The percentile that all features above it will be filtered out.
column (str) – Name of the DataFrame column according to which the filtering will be performed.
interpolate ('nearest', 'higher', 'lower', 'midpoint' or 'linear' (default='linear')) – interpolation method to use when the desired quantile lies between two data points.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # keep only the rows whose value in the column 'log2FoldChange' is below the 75th percentile >>> d.filter_percentile(0.75,'log2FoldChange') Filtered 7 features, leaving 21 of the original 28 features. Filtered inplace.
>>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # keep only the rows vulse value in the column 'log2FoldChange' is above the 25th percentile >>> d.filter_percentile(0.25,'log2FoldChange',opposite=True) Filtered 7 features, leaving 21 of the original 28 features. Filtered inplace.
- filter_top_n(by: ColumnNames, n: PositiveInt = 100, ascending: bool | List[bool] = True, na_position: str = 'last', opposite: bool = False, inplace: bool = True)
Sort the rows by the values of specified column or columns, then keep only the top ‘n’ rows.
- Parameters:
by (name of column/columns (str/List[str])) – Names of the column or columns to sort and then filter by.
n (int) – How many features to keep in the Filter object.
ascending (bool or list of bools (default=True)) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of Filter.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only the 10 rows with the highest values in the columns 'cond1' >>> counts.filter_top_n(by='cond1',n=10, ascending=False) Filtered 12 features, leaving 10 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only the 10 rows which have the lowest values in the columns 'cond1' >>> # and then the highest values in the column 'cond2' >>> counts.filter_top_n(by=['cond1','cond2'],n=10, ascending=[True,False]) Filtered 12 features, leaving 10 of the original 22 features. Filtered inplace.
- find_paralogs_ensembl(organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True)
Find paralogs within the same species using the Ensembl database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
- Returns:
DataFrame describing all discovered paralog mappings.
- find_paralogs_panther(organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto')
Find paralogs within the same species using the PantherDB database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- Returns:
DataFrame describing all discovered paralog mappings.
- fname
filename with full path
- head(n: PositiveInt = 5) DataFrame
Return the first n rows of the Filter object. See polars.DataFrame.head documentation.
- Parameters:
n (positive int, default 5) – Number of rows to show.
- Returns:
returns the first n rows of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> d.head() shape: (5, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬───────────┬─────────────┬─────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪═══════════╪═════════════╪═════════════╡ │ WBGene00000002 ┆ 6820.755327 ┆ 7.567762 ┆ 0.142257 ┆ 53.197692 ┆ 0.0 ┆ 0.0 │ │ WBGene00000003 ┆ 3049.62567 ┆ 9.138071 ┆ 0.245989 ┆ 37.148359 ┆ 4.6600e-302 ┆ 4.2800e-298 │ │ WBGene00000004 ┆ 1432.911791 ┆ 8.111737 ┆ 0.246801 ┆ 32.867521 ┆ 6.4000e-237 ┆ 3.9200e-233 │ │ WBGene00000005 ┆ 4028.154186 ┆ 6.534112 ┆ 0.202467 ┆ 32.272543 ┆ 1.7000e-228 ┆ 7.8000e-225 │ │ WBGene00000006 ┆ 1230.58524 ┆ 7.157428 ┆ 0.227948 ┆ 31.399325 ┆ 2.0700e-216 ┆ 7.5900e-213 │ └────────────────┴─────────────┴────────────────┴──────────┴───────────┴─────────────┴─────────────┘
>>> d.head(3) # return only the first 3 rows shape: (3, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬───────────┬─────────────┬─────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪═══════════╪═════════════╪═════════════╡ │ WBGene00000002 ┆ 6820.755327 ┆ 7.567762 ┆ 0.142257 ┆ 53.197692 ┆ 0.0 ┆ 0.0 │ │ WBGene00000003 ┆ 3049.62567 ┆ 9.138071 ┆ 0.245989 ┆ 37.148359 ┆ 4.6600e-302 ┆ 4.2800e-298 │ │ WBGene00000004 ┆ 1432.911791 ┆ 8.111737 ┆ 0.246801 ┆ 32.867521 ┆ 6.4000e-237 ┆ 3.9200e-233 │ └────────────────┴─────────────┴────────────────┴──────────┴───────────┴─────────────┴─────────────┘
- property index_set: set
Returns all of the features in the current DataFrame (which were not removed by previously used filter methods) as a set. if any duplicate features exist in the filter object (same WBGene appears more than once), the corresponding WBGene index will appear in the returned set ONLY ONCE.
- Returns:
A set of WBGene names.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> myset = counts.index_set >>> print(myset) {'WBGene00044022', 'WBGene00077504', 'WBGene00007079', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00077503', 'WBGene00007078', 'WBGene00007064', 'WBGene00077502', 'WBGene00044951', 'WBGene00007077', 'WBGene00007066', 'WBGene00007076', 'WBGene00014997', 'WBGene00043990', 'WBGene00007074', 'WBGene00043987', 'WBGene00007071', 'WBGene00043989', 'WBGene00043988', 'WBGene00007075'}
- property index_string: str
Returns a string of all feature indices in the current DataFrame, sorted by their current order in the FIlter object, and separated by newline.
- This includes all of the feature indices which were not filtered out by previously-used filter methods.
if any duplicate features exist in the filter object (same index appears more than once),
the corresponding index will appear in the returned string ONLY ONCE.
- Returns:
A string of WBGene indices separated by newlines (\n).
For example, “WBGene00000001\nWBGene00000003\nWBGene12345678”.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.sort(by='cond1',ascending=False) >>> mystring = counts.index_string >>> print(mystring) WBGene00007075 WBGene00043988 WBGene00043990 WBGene00007079 WBGene00007076 WBGene00043989 WBGene00007063 WBGene00007077 WBGene00007078 WBGene00007071 WBGene00007064 WBGene00007066 WBGene00007074 WBGene00043987 WBGene00007067 WBGene00014997 WBGene00044022 WBGene00077503 WBGene00077504 WBGene00077502 WBGene00007069 WBGene00044951
- intersection(*others: Filter | set, return_type: Literal['set', 'str'] = 'set', inplace: bool = False)
Keep only the features that exist in ALL of the given Filter objects/sets. Can be done either inplace on the first Filter object, or return a set/string of features.
- Parameters:
others (Filter or set objects.) – Objects to calculate intersection with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the intersecting features. If ‘str’, returns a string of the intersecting features, delimited by a comma.
inplace (bool (default=False)) – If True, the function will be applied in-place to the current Filter object. If False (default), the function will return a set/str that contains the intersecting indices.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that intersect between the given Filter objects/sets.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> # calculate intersection and return a set >>> d.intersection(a_set) {'WBGene00000002', 'WBGene00000003'}
# calculate intersection and filter in-place >>> d.intersection(a_set, inplace=True) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
- majority_vote_intersection(*others: Filter | set, majority_threshold: float = 0.5, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the features that appear in at least (majority_threhold * 100)% of the given Filter objects/sets. Majority-vote intersection with majority_threshold=0 is equivalent to Union. Majority-vote intersection with majority_threshold=1 is equivalent to Intersection.
- Parameters:
others (Filter or set objects.) – Objects to calculate intersection with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the intersecting WBGene indices. If ‘str’, returns a string of the intersecting indices, delimited by a comma.
majority_threshold (float (default=0.5)) – The threshold that determines what counts as majority. Features will be returned only if they appear in at least (majority_threshold * 100)% of the given Filter objects/sets.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that uphold majority vote intersection between two given Filter objects/sets.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> b_set = {'WBGene00000002','WBGene00000004'} >>> # calculate majority-vote intersection and return a set >>> d.majority_vote_intersection(a_set, b_set, majority_threshold=2/3) {'WBGene00000002', 'WBGene00000003', 'WBGene00000004'}
- map_orthologs_ensembl(map_to_organism: str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'], map_from_organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the Ensembl database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_orthoinspector(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the OrthoInspector database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_panther(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_least_diverged: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PantherDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_least_diverged (bool (default=True)) – if True (default), RNAlysis will only fetch ortholog mappings that were flagged as a ‘least diverged ortholog’ on the PantherDB database. You can read more about this flag on the PantherDB website: https://www.pantherdb.org/genes/
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_phylomedb(map_to_organism: str | int | Literal, map_from_organism: Literal['auto'] | str | int | Literal = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', consistency_score_threshold: Fraction = 0.5, filter_consistency_score: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PhylomeDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- param map_to_organism:
organism name or NCBI taxon ID of the target species for ortholog mapping.
- type map_to_organism:
str or int
- param map_from_organism:
organism name or NCBI taxon ID of the input genes’ source species.
- type map_from_organism:
str or int
- param gene_id_type:
the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- type gene_id_type:
str or ‘auto’ (default=’auto’)
- ` :param consistency_score_threshold: the minimum consistency score required for an ortholog mapping to be considered valid. Consistency scores are calculated by PhylomeDB and represent the confidence of the ortholog mapping. setting consistency_score_threshold to 0 will keep all mappings. You can read more about PhylomeDB consistency score on the PhylomeDB website: orthology.phylomedb.org/help
- type consistency_score_threshold:
float between 0 and 1 (default=0.5)
- param filter_consistency_score:
if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest consistency score.
- type filter_consistency_score:
bool (default=True)
- param non_unique_mode:
How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
- type non_unique_mode:
‘first’, ‘last’, ‘random’, or ‘none’ (default=’first’)
- param remove_unmapped_genes:
if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
- type remove_unmapped_genes:
bool (default=False)
- type inplace:
bool (default=True)
- param inplace:
If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- return:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- number_filters(column: ColumnName, operator: Literal['greater than', 'equals', 'lesser than', 'abs greater than'], value: float, opposite: bool = False, inplace: bool = True)
Applay a number filter (greater than, equal, lesser than) on a particular column in the Filter object.
- Parameters:
column (str) – name of the column to filter by
operator (str: 'gt' / 'greater than' / '>', 'eq' / 'equals' / '=', 'lt' / 'lesser than' / '<') – the operator to filter the column by (greater than, equal or lesser than)
value (float) – the value to filter by
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','gt',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
>>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','greater than',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
>>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','>',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
- print_features()
Print the feature indices in the Filter object, sorted by their current order in the FIlter object, and separated by newline.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.sort(by='cond1',ascending=False) >>> counts.print_features() WBGene00007075 WBGene00043988 WBGene00043990 WBGene00007079 WBGene00007076 WBGene00043989 WBGene00007063 WBGene00007077 WBGene00007078 WBGene00007071 WBGene00007064 WBGene00007066 WBGene00007074 WBGene00043987 WBGene00007067 WBGene00014997 WBGene00044022 WBGene00077503 WBGene00077504 WBGene00077502 WBGene00007069 WBGene00044951
- save_csv(alt_filename: None | str | Path = None)
Saves the current filtered data to a .csv file.
- Parameters:
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- save_parquet(alt_filename: None | str | Path = None)
Saves the current filtered data to a .parquet file.
- Parameters:
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- save_table(suffix: Literal['.csv', '.tsv', '.parquet'] = '.csv', alt_filename: None | str | Path = None)
Save the current filtered data table.
- Parameters:
suffix ('.csv', '.tsv', or '.parquet' (default='.csv')) – the file suffix
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- sort(by: str | List[str], ascending: bool | List[bool] = True, na_position: str = 'last', inplace: bool = True)
Sort the rows by the values of specified column or columns.
- Parameters:
by (str or list of str) – Names of the column or columns to sort by.
ascending (bool or list of bool (default=True)) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
inplace (bool (default=True)) – If True, perform operation in-place. Otherwise, returns a sorted copy of the Filter object without modifying the original.
- Returns:
None if inplace=True, a sorted Filter object otherwise.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.head() cond1 cond2 cond3 cond4 WBGene00007063 633 451 365 388 WBGene00007064 60 57 20 23 WBGene00044951 0 0 0 1 WBGene00007066 55 266 46 39 WBGene00007067 15 13 1 0 >>> counts.sort(by='cond1',ascending=True) >>> counts.head() cond1 cond2 cond3 cond4 WBGene00044951 0 0 0 1 WBGene00077504 0 0 0 0 WBGene00007069 0 2 1 0 WBGene00077502 0 0 0 0 WBGene00077503 1 4 2 0
- split_by_attribute(attributes: str | List[str], ref: str | Path | Literal['predefined'] = 'predefined') tuple
Splits the features in the Filter object into multiple Filter objects, each corresponding to one of the specified Attribute Reference Table attributes. Each new Filter object will contain only features that belong to its Attribute Reference Table attribute.
- Parameters:
attributes (list of strings) – list of attribute names from the Attribute Reference Table to filter by.
ref – filename/path of the reference table to be used as reference.
- Return type:
Tuple[filtering.Filter]
- Returns:
A tuple of Filter objects, each containing only features that match one Attribute Reference Table attribute; the Filter objects are returned in the same order the attributes were given in.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> attribute1,attribute2 = counts.split_by_attribute(['attribute1','attribute2'], ... ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 15 features, leaving 7 of the original 22 features. Filtering result saved to new object. Filtered 20 features, leaving 2 of the original 22 features. Filtering result saved to new object.
- split_by_percentile(percentile: Fraction, column: ColumnName, interpolate: 'nearest', 'higher', 'lower', 'midpoint', 'linear' = 'linear') tuple
Splits the features in the Filter object into two non-overlapping Filter objects: one containing features below the specified percentile in the specfieid column, and the other containing features about the specified percentile in the specified column.
- Parameters:
percentile (float between 0 and 1) – The percentile that all features above it will be filtered out.
column (str) – Name of the DataFrame column according to which the filtering will be performed.
- Return type:
Tuple[filtering.Filter, filtering.Filter]
- Returns:
a tuple of two Filter objects: the first contains all of the features below the specified percentile, and the second contains all of the features above and equal to the specified percentile.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> below, above = d.split_by_percentile(0.75,'log2FoldChange') Filtered 7 features, leaving 21 of the original 28 features. Filtering result saved to new object. Filtered 21 features, leaving 7 of the original 28 features. Filtering result saved to new object.
- symmetric_difference(other: Filter | set, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the WBGene indices that exist either in the first Filter object/set OR the second, but NOT in both (set symmetric difference).
- Parameters:
other (Filter or set.) – a second Filter object/set to calculate symmetric difference with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the features that exist in exactly one Filter object. If ‘str’, returns a string of the features that exist in exactly one Filter object, delimited by a comma.
- Return type:
set or str
- Returns:
a set/string of the features that that exist t in exactly one Filter. (set symmetric difference).
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter("tests/test_files/test_deseq.csv") >>> counts = filtering.CountFilter('tests/test_files/counted.csv') >>> # calculate difference and return a set >>> d.symmetric_difference(counts) {'WBGene00000017', 'WBGene00077504', 'WBGene00000024', 'WBGene00000010', 'WBGene00000020', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00007078', 'WBGene00000029', 'WBGene00000006', 'WBGene00007064', 'WBGene00000019', 'WBGene00000004', 'WBGene00007066', 'WBGene00014997', 'WBGene00000023', 'WBGene00007074', 'WBGene00000025', 'WBGene00043989', 'WBGene00043988', 'WBGene00000014', 'WBGene00000027', 'WBGene00000021', 'WBGene00044022', 'WBGene00007079', 'WBGene00000012', 'WBGene00000005', 'WBGene00077503', 'WBGene00000026', 'WBGene00000003', 'WBGene00000002', 'WBGene00077502', 'WBGene00044951', 'WBGene00007077', 'WBGene00000007', 'WBGene00000008', 'WBGene00007076', 'WBGene00000013', 'WBGene00043990', 'WBGene00043987', 'WBGene00007071', 'WBGene00000011', 'WBGene00000015', 'WBGene00000018', 'WBGene00000016', 'WBGene00000028', 'WBGene00007075', 'WBGene00000022', 'WBGene00000009'}
- tail(n: PositiveInt = 5) DataFrame
Return the last n rows of the Filter object. See polars.DataFrame.tail documentation.
- Parameters:
n (positive int, default 5) – Number of rows to show.
- Return type:
pl.DataFrame
- Returns:
returns the last n rows of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> d.tail() shape: (5, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬────────────┬────────────┬────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪════════════╪════════════╪════════════╡ │ WBGene00000025 ┆ 2236.185837 ┆ 2.477374 ┆ 0.129606 ┆ 19.114626 ┆ 1.9100e-81 ┆ 1.4600e-78 │ │ WBGene00000026 ┆ 343.648987 ┆ -4.037191 ┆ 0.219781 ┆ -18.369115 ┆ 2.3200e-75 ┆ 1.7000e-72 │ │ WBGene00000027 ┆ 175.142856 ┆ 6.352044 ┆ 0.347777 ┆ 18.264706 ┆ 1.5800e-74 ┆ 1.1200e-71 │ │ WBGene00000028 ┆ 219.1632 ┆ 3.913657 ┆ 0.217802 ┆ 17.968851 ┆ 3.4200e-72 ┆ 2.3200e-69 │ │ WBGene00000029 ┆ 1066.242402 ┆ -2.811281 ┆ 0.158284 ┆ -17.761002 ┆ 1.4200e-70 ┆ 9.2900e-68 │ └────────────────┴─────────────┴────────────────┴──────────┴────────────┴────────────┴────────────┘
>>> d.tail(8) # returns the last 8 rows shape: (8, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬────────────┬────────────┬────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪════════════╪════════════╪════════════╡ │ WBGene00000022 ┆ 365.813048 ┆ 6.101303 ┆ 0.291484 ┆ 20.931891 ┆ 2.7400e-97 ┆ 2.4000e-94 │ │ WBGene00000023 ┆ 3168.566714 ┆ 3.906719 ┆ 0.190439 ┆ 20.514331 ┆ 1.6000e-93 ┆ 1.3400e-90 │ │ WBGene00000024 ┆ 221.925724 ┆ 4.801676 ┆ 0.246313 ┆ 19.494189 ┆ 1.2300e-84 ┆ 9.8200e-82 │ │ WBGene00000025 ┆ 2236.185837 ┆ 2.477374 ┆ 0.129606 ┆ 19.114626 ┆ 1.9100e-81 ┆ 1.4600e-78 │ │ WBGene00000026 ┆ 343.648987 ┆ -4.037191 ┆ 0.219781 ┆ -18.369115 ┆ 2.3200e-75 ┆ 1.7000e-72 │ │ WBGene00000027 ┆ 175.142856 ┆ 6.352044 ┆ 0.347777 ┆ 18.264706 ┆ 1.5800e-74 ┆ 1.1200e-71 │ │ WBGene00000028 ┆ 219.1632 ┆ 3.913657 ┆ 0.217802 ┆ 17.968851 ┆ 3.4200e-72 ┆ 2.3200e-69 │ │ WBGene00000029 ┆ 1066.242402 ┆ -2.811281 ┆ 0.158284 ┆ -17.761002 ┆ 1.4200e-70 ┆ 9.2900e-68 │ └────────────────┴─────────────┴────────────────┴──────────┴────────────┴────────────┴────────────┘
- text_filters(column: ColumnName, operator: Literal['equals', 'contains', 'starts with', 'ends with'], value: str, opposite: bool = False, inplace: bool = True)
Applay a text filter (equals, contains, starts with, ends with) on a particular column in the Filter object.
- Parameters:
column (str) – name of the column to filter by
operator (str: 'eq' / 'equals' / '=', 'ct' / 'contains' / 'in', 'sw' / 'starts with', 'ew' / 'ends with') – the operator to filter the column by (equals, contains, starts with, ends with)
value (number (int or float)) – the value to filter by
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/text_filters.csv') >>> # keep only rows that have a value that starts with 'AC3' in the column 'name'. >>> filt.text_filters('name','sw','AC3') Filtered 17 features, leaving 5 of the original 22 features. Filtered inplace.
- transform(function: Literal['Box-Cox', 'log2', 'log10', 'ln', 'Standardize'] | Callable, columns: ColumnNames | Literal['all'] = 'all', inplace: bool = True, **function_kwargs)
Transform the values in the Filter object with the specified function.
- Parameters:
function (Callable or str ('logx' for base-x log of the data + 1, 'box-cox' for Box-Cox transform of the data + 1, 'standardize' for standardization)) – The function or function name to be applied.
columns (str, list of str, or 'all' (default='all')) – The columns to which the transform should be applied.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
function_kwargs – Any additional keyworded arguments taken by the supplied function.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/counted.csv') >>> filt_log10 = filt.transform('log10', inplace=False) Transformed 22 features. Transformation result saved to new object. >>> filt.transform(lambda x: x+1, columns=['cond1','cond4']) Transformed 22 features. Transformed inplace.
- translate_gene_ids(translate_to: str | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'], translate_from: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', remove_unmapped_genes: bool = False, inplace: bool = True)
Translates gene names/IDs from one type to another. Mapping is done using the UniProtKB Gene ID Mapping service. You can choose to optionally drop from the table all rows that failed to be translated.
- Parameters:
translate_to (str) – the gene ID type to translate gene names/IDs to. For example: UniProtKB, Ensembl, Wormbase.
translate_from (str or 'auto' (default='auto')) – the gene ID type to translate gene names/IDs from. For example: UniProtKB, Ensembl, Wormbase. If translate_from=’auto’, RNAlysis will attempt to automatically determine the gene ID type of the features in the table.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be translated will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and translated instance of the Filter object.
- union(*others: Filter | set, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the union of features between multiple Filter objects/sets (the features that exist in at least one of the Filter objects/sets).
- Parameters:
others (Filter or set objects.) – Objects to calculate union with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the union features. If ‘str’, returns a string of the union WBGene indices, delimited by a comma.
- Return type:
set or str
- Returns:
a set/string of the WBGene indices that exist in at least one of the Filter objects.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # calculate union and return a set >>> d.union(counts) {'WBGene00000017', 'WBGene00000021', 'WBGene00044022', 'WBGene00077504', 'WBGene00000012', 'WBGene00000024', 'WBGene00007079', 'WBGene00000010', 'WBGene00000020', 'WBGene00000005', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00077503', 'WBGene00007078', 'WBGene00000026', 'WBGene00000029', 'WBGene00000002', 'WBGene00000003', 'WBGene00000006', 'WBGene00007064', 'WBGene00077502', 'WBGene00044951', 'WBGene00000007', 'WBGene00000008', 'WBGene00000019', 'WBGene00007077', 'WBGene00000004', 'WBGene00007066', 'WBGene00007076', 'WBGene00000013', 'WBGene00014997', 'WBGene00000023', 'WBGene00043990', 'WBGene00007074', 'WBGene00000025', 'WBGene00000011', 'WBGene00043987', 'WBGene00007071', 'WBGene00000015', 'WBGene00000018', 'WBGene00043989', 'WBGene00043988', 'WBGene00000014', 'WBGene00000016', 'WBGene00000027', 'WBGene00000028', 'WBGene00007075', 'WBGene00000022', 'WBGene00000009'}
- class rnalysis.filtering.FoldChangeFilter(fname: str | Path | tuple, numerator_name: str, denominator_name: str, suppress_warnings: bool = False)
Bases:
Filter- A class that contains a single column, representing the gene-specific fold change between two conditions.
this class does not support ‘inf’ and ‘0’ values, and importing a file with such values could lead to incorrect filtering and statistical analyses.
Attributes
- df: polars DataFrame
A DataFrame containing the gene names and their fold change values. The DataFrame is modified upon usage of filter operations.
- shape: tuple (rows, columns)
The dimensions of df.
- columns: list
The columns of df.
- fname: pathlib.Path
The path and filename for the purpose of saving df as a csv file. Updates automatically when filter operations are applied.
- index_set: set
All of the indices in the current DataFrame (which were not removed by previously used filter methods) as a set.
- index_string: string
A string of all feature indices in the current DataFrame separated by newline.
- numerator: str
Name of the numerator used to calculate the fold change.
- denominator: str
Name of the denominator used to calculate the fold change.
- static _from_string(msg: str = '', delimiter: str = '\n')
Takes a manual string input from the user, and then splits it using a delimiter into a list of values.
- param msg:
a promprt to be printed to the user
- param delimiter:
the delimiter used to separate the values. Default is ‘
- ‘
- return:
A list of the comma-seperated values the user inserted.
- _inplace(new_df: DataFrame, opposite: bool, inplace: bool, suffix: str, printout_operation: str = 'filter', **filter_update_kwargs)
Executes the user’s choice whether to filter in-place or create a new instance of the Filter object.
- Parameters:
new_df (pl.DataFrame) – the post-filtering DataFrame
opposite (bool) – Determines whether to return the filtration ,or its opposite.
inplace (bool) – Determines whether to filter in-place or not.
suffix (str) – The suffix to be added to the filename
- Returns:
If inplace is False, returns a new instance of the Filter object.
- _set_ops(others, return_type: Literal['set', 'str'], op: Any, **kwargs)
Apply the supplied set operation (union/intersection/difference/symmetric difference) to the supplied objects.
- Parameters:
others (Filter or set objects.) – the other objects to apply the set operation to
return_type ('set' or 'str') – the return type of the output
op (function (set.union, set.intersection, set.difference or set.symmetric_difference)) – the set operation
kwargs – any additional keyworded arguments to be supplied to the set operation.
- Returns:
a set/string of indices resulting from the set operation
- Return type:
set or str
- _sort(by: str | List[str], ascending: bool | List[bool] = True, na_position: str = 'last')
Sort the rows by the values of specified column or columns.
- Parameters:
by (str or list of str) – Names of the column or columns to sort by.
ascending (bool or list of bool, default True) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
inplace (bool, default True) – If True, perform operation in-place. Otherwise, returns a sorted copy of the Filter object without modifying the original.
- Returns:
None if inplace=True, a sorted Filter object otherwise.
- annotate_from_gtf(gtf_path: str | Path, attribute: Literal['gene_biotype', 'transcript_biotype', 'biotype', 'gene_name', 'gene_id', 'transcript_id', 'chromosome', 'source', 'strand'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', column_name: str | None = None, inplace: bool = True)
Adds a new column to the table, annotating each feature with the value of a GTF/GFF attribute (for example: the biotype, chromosome, strand, or source of each gene). Features that are not found in the annotation file are annotated with a missing value.
- Parameters:
gtf_path (str or Path) – Path to your GTF/GFF annotation file. The file should match the type of gene names/IDs you use in your table.
attribute (str (default='gene_biotype')) – name of the attribute to annotate with. Standard column-9 attributes (such as ‘gene_biotype’, ‘gene_name’, or any custom key in your file) are supported, as well as the reserved names ‘chromosome’, ‘source’ and ‘strand’, which are read from the fixed columns of the annotation file.
feature_type ('gene' or 'transcript' (default='gene')) – determines whether the features/rows in your data table describe individual genes or transcripts.
column_name (str or None (default=None)) – the name of the new annotation column. If not specified, the attribute name is used. If a column with this name already exists in the table, it will be overwritten.
inplace (bool (default=True)) – If True (default), the annotation column will be added to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and annotated instance of the Filter object.
- biotypes_from_gtf(gtf_path: str | Path, attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', long_format: bool = False) DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- biotypes_from_ref_table(long_format: bool = False, ref: str | Path | Literal['predefined'] = 'predefined') DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :param ref: Name of the biotype reference table used to determine biotype. Default is ce11 (included in the package). :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # short-form view >>> d.biotypes_from_ref_table(ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 2) ┌────────────────┬───────┐ │ biotype ┆ count │ │ --- ┆ --- │ │ str ┆ u32 │ ╞════════════════╪═══════╡ │ unknown ┆ 1 │ │ protein_coding ┆ 26 │ │ pseudogene ┆ 1 │ └────────────────┴───────┘
>>> # long-form view >>> d.biotypes_from_ref_table(long_format=True,ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 49) ┌───────────┬───────────┬───────────┬───────────┬───┬───────────┬───────────┬───────────┬──────────┐ │ biotype ┆ baseMean_ ┆ baseMean_ ┆ baseMean_ ┆ … ┆ padj_25% ┆ padj_50% ┆ padj_75% ┆ padj_max │ │ --- ┆ count ┆ mean ┆ std ┆ ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ --- ┆ --- ┆ --- ┆ ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ │ ┆ f64 ┆ f64 ┆ f64 ┆ ┆ ┆ ┆ ┆ │ ╞═══════════╪═══════════╪═══════════╪═══════════╪═══╪═══════════╪═══════════╪═══════════╪══════════╡ │ protein_c ┆ 26.0 ┆ 1823.0896 ┆ 1796.5207 ┆ … ┆ 3.8506e-1 ┆ 1.1350e-1 ┆ 1.0051e-9 ┆ 9.2900e- │ │ oding ┆ ┆ 09 ┆ 8 ┆ ┆ 76 ┆ 35 ┆ 0 ┆ 68 │ │ pseudogen ┆ 1.0 ┆ 2688.0437 ┆ null ┆ … ┆ 1.8000e-9 ┆ 1.8000e-9 ┆ 1.8000e-9 ┆ 1.8000e- │ │ e ┆ ┆ 01 ┆ ┆ ┆ 4 ┆ 4 ┆ 4 ┆ 94 │ │ unknown ┆ 1.0 ┆ 2085.9950 ┆ null ┆ … ┆ 3.0700e-1 ┆ 3.0700e-1 ┆ 3.0700e-1 ┆ 3.0700e- │ │ ┆ ┆ 94 ┆ ┆ ┆ 52 ┆ 52 ┆ 52 ┆ 152 │ └───────────┴───────────┴───────────┴───────────┴───┴───────────┴───────────┴───────────┴──────────┘
- property columns: list
The columns of df.
- Returns:
a list of the columns in the Filter object.
- Return type:
list
- denominator
name of the denominator
- describe(percentiles: float | List[float] = (0.01, 0.25, 0.5, 0.75, 0.99)) DataFrame
Generate descriptive statistics that summarize the central tendency, dispersion and shape of the dataset’s distribution, excluding NaN values. For more information see the documentation of polars.DataFrame.describe.
- Parameters:
percentiles (list-like of floats (default=(0.01, 0.25, 0.5, 0.75, 0.99))) – The percentiles to include in the output. All should fall between 0 and 1. The default is [.25, .5, .75], which returns the 25th, 50th, and 75th percentiles.
- Returns:
Summary statistics of the dataset.
- Return type:
pl.DataFrame
- Examples:
>>> from rnalysis import filtering >>> import numpy as np >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.describe() cond1 cond2 cond3 cond4 count 22.000000 22.000000 22.000000 22.000000 mean 2515.590909 2209.227273 4230.227273 3099.818182 std 4820.512674 4134.948493 7635.832664 5520.394522 min 0.000000 0.000000 0.000000 0.000000 1% 0.000000 0.000000 0.000000 0.000000 25% 6.000000 6.250000 1.250000 0.250000 50% 57.500000 52.500000 23.500000 21.000000 75% 2637.000000 2479.000000 6030.500000 4669.750000 99% 15054.950000 12714.290000 21955.390000 15603.510000 max 15056.000000 12746.000000 22027.000000 15639.000000
>>> # show the deciles (10%, 20%, 30%... 90%) of the columns >>> counts.describe(percentiles=np.arange(0.1, 1, 0.1)) cond1 cond2 cond3 cond4 count 22.000000 22.000000 22.000000 22.000000 mean 2515.590909 2209.227273 4230.227273 3099.818182 std 4820.512674 4134.948493 7635.832664 5520.394522 min 0.000000 0.000000 0.000000 0.000000 10% 0.000000 0.200000 0.000000 0.000000 20% 1.400000 3.200000 1.000000 0.000000 30% 15.000000 15.700000 2.600000 1.000000 40% 28.400000 26.800000 14.000000 9.000000 50% 57.500000 52.500000 23.500000 21.000000 60% 82.000000 106.800000 44.000000 33.000000 70% 484.200000 395.500000 305.000000 302.500000 80% 3398.600000 3172.600000 7981.400000 6213.000000 90% 8722.100000 7941.800000 16449.500000 12129.900000 max 15056.000000 12746.000000 22027.000000 15639.000000
- df
- difference(*others: Filter | set, return_type: Literal['set', 'str'] = 'set', inplace: bool = False)
Keep only the features that exist in the first Filter object/set but NOT in the others. Can be done inplace on the first Filter object, or return a set/string of features.
- Parameters:
others (Filter or set objects.) – Objects to calculate difference with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the features that exist only in the first Filter object. If ‘str’, returns a string of the WBGene indices that exist only in the first Filter object, delimited by a comma.
inplace (bool, default False) – If True, filtering will be applied to the current Filter object. If False (default), the function will return a set/str that contains the intersecting features.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that exist only in the first Filter object/set (set difference).
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter("tests/test_files/test_deseq.csv") >>> counts = filtering.CountFilter('tests/test_files/counted.csv') >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> # calculate difference and return a set >>> d.difference(counts, a_set) {'WBGene00007063', 'WBGene00007064', 'WBGene00007066', 'WBGene00007067', 'WBGene00007069', 'WBGene00007071', 'WBGene00007074', 'WBGene00007075', 'WBGene00007076', 'WBGene00007077', 'WBGene00007078', 'WBGene00007079', 'WBGene00014997', 'WBGene00043987', 'WBGene00043988', 'WBGene00043989', 'WBGene00043990', 'WBGene00044022', 'WBGene00044951', 'WBGene00077502', 'WBGene00077503', 'WBGene00077504'}
# calculate difference and filter in-place >>> d.difference(counts, a_set, inplace=True) Filtered 2 features, leaving 26 of the original 28 features. Filtered inplace.
- drop_columns(columns: ColumnNames, inplace: bool = True)
Drop specific columns from the table.
- Parameters:
columns (str or list of str) – The names of the column/columns to be dropped fro mthe table.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_abs_log2_fold_change(abslog2fc: float = 1, opposite: bool = False, inplace: bool = True)
Filters out all features whose absolute log2 fold change is below the indicated threshold. For example: if log2fc is 1.0, all features whose log2 fold change is between 1 and -1 (went up less than two-fold or went down less than two-fold) will be filtered out.
- Parameters:
abslog2fc – The threshold absolute log2 fold change for filtering out a feature. Float or int. All features whose absolute log2 fold change is lower than log2fc will be filtered out.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current FoldChangeFilter object. If False, the function will return a new FoldChangeFilter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of FoldChangeFilter.
- Examples:
>>> from rnalysis import filtering >>> f = filtering.FoldChangeFilter('tests/test_files/fc_1.csv','numerator name','denominator name') >>> f.filter_abs_log2_fold_change(2) # keep only rows whose log2(fold change) is >=2 or <=-2 Filtered 18 features, leaving 4 of the original 22 features. Filtered inplace.
- filter_biotype_from_gtf(gtf_path: str | Path, biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
biotype (str or list of strings) – the biotypes which will not be filtered out.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- filter_biotype_from_ref_table(biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
- Parameters:
biotype (string or list of strings) – the biotypes which will not be filtered out.
ref – Name of the biotype reference file used to determine biotypes. Default is the path defined by the user in the settings.yaml file.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows whose biotype is 'protein_coding' >>> counts.filter_biotype_from_ref_table('protein_coding',ref='tests/test_files/biotype_ref_table_for_tests.csv') Filtered 9 features, leaving 13 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows whose biotype is 'protein_coding' or 'pseudogene' >>> counts.filter_biotype_from_ref_table(['protein_coding','pseudogene'],ref='tests/test_files/biotype_ref_table_for_tests.csv') Filtered 0 features, leaving 22 of the original 22 features. Filtered inplace.
- filter_by_attribute(attributes: str | List[str] = None, mode: Literal['union', 'intersection'] = 'union', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters features according to user-defined attributes from an Attribute Reference Table. When multiple attributes are given, filtering can be done in ‘union’ mode (where features that belong to at least one attribute are not filtered out), or in ‘intersection’ mode (where only features that belong to ALL attributes are not filtered out). To learn more about user-defined attributes and Attribute Reference Tables, read the user guide.
- Parameters:
attributes (string or list of strings, which are column titles in the user-defined Attribute Reference Table.) – attributes to filter by.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
ref (str or pathlib.Path (default='predefined')) – filename/path of the attribute reference table to be used as reference.
opposite (bool (default=False)) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to the attribute 'attribute1' >>> counts.filter_by_attribute('attribute1',ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 15 features, leaving 7 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to the attributes 'attribute1' OR 'attribute3' (union) >>> counts.filter_by_attribute(['attribute1','attribute3'],ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 14 features, leaving 8 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that belong to both attributes 'attribute1' AND 'attribute3' (intersection) >>> counts.filter_by_attribute(['attribute1','attribute3'],mode='intersection', ... ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 19 features, leaving 3 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that DON'T belong to either 'attribute1','attribute3' or both >>> counts.filter_by_attribute(['attribute1','attribute3'],ref='tests/test_files/attr_ref_table_for_examples.csv', ... opposite=True) Filtered 8 features, leaving 14 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only rows that DON'T belong to both 'attribute1' AND 'attribute3' >>> counts.filter_by_attribute(['attribute1','attribute3'],mode='intersection', ... ref='tests/test_files/attr_ref_table_for_examples.csv',opposite=True) Filtered 3 features, leaving 19 of the original 22 features. Filtered inplace.
- filter_by_go_annotations(go_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', propagate_annotations: bool = True, evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', opposite: bool = False, inplace: bool = True)
Filters genes according to GO annotations, keeping only genes that are annotated with a specific GO term. When multiple GO terms are given, filtering can be done in ‘union’ mode (where genes that belong to at least one GO term are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL GO terms are not filtered out).
- Parameters:
go_ids (str or list of str)
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :param propagate_annotations: determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/ :type propagate_annotations: ‘classic’, ‘elim’, ‘weight’, ‘all.m’, or ‘no’ (default=’elim’) :param evidence_types: only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or ‘any’ (default=’any’) :param excluded_evidence_types: annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type excluded_evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or None (default=None) :param databases: only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current FeatureSet object. If False, the function will return a new FeatureSet instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the FeatureSet object.
- filter_by_gtf_attribute(gtf_path: str | Path, attribute: Literal['gene_biotype', 'transcript_biotype', 'biotype', 'gene_name', 'gene_id', 'transcript_id', 'chromosome', 'source', 'strand'] | str = 'gene_biotype', value: str | List[str] = 'protein_coding', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters the features in the table by any attribute described in a GTF/GFF annotation file, keeping only features whose attribute matches one of the specified values (for example: keep only genes on a specific chromosome or strand, from a specific source, or of a specific biotype). This is a generalization of filter_biotype_from_gtf to any GTF/GFF attribute.
- Parameters:
gtf_path (str or Path) – Path to your GTF/GFF annotation file. The file should match the type of gene names/IDs you use in your table.
attribute (str (default='gene_biotype')) – name of the attribute to filter by. Standard column-9 attributes (such as ‘gene_biotype’, ‘gene_name’, or any custom key in your file) are supported, as well as the reserved names ‘chromosome’, ‘source’ and ‘strand’, which are read from the fixed columns of the annotation file.
value (str or list of strings) – the attribute value/values which will NOT be filtered out. For example, to keep only the features on chromosomes ‘chr1’ and ‘chr2’, set attribute=’chromosome’ and value=[‘chr1’, ‘chr2’].
feature_type ('gene' or 'transcript' (default='gene')) – determines whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- filter_by_kegg_annotations(kegg_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', opposite: bool = False, inplace: bool = True)
Filters genes according to KEGG pathways, keeping only genes that belong to specific KEGG pathway. When multiple KEGG IDs are given, filtering can be done in ‘union’ mode (where genes that belong to at least one pathway are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL pathways are not filtered out).
- Parameters:
kegg_ids (str or list of str) – the KEGG pathway IDs according to which the table will be filtered. An example for a legal KEGG pathway ID would be ‘path:cel04020’ for the C. elegans calcium signaling pathway.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the Filter object.
- filter_by_row_name(row_names: str | List[str], opposite: bool = False, inplace: bool = True)
Filter out specific rows from the table by their name (index).
- Parameters:
row_names (str or list of str) – list of row names to be removed from the table.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_duplicate_ids(keep: Literal['first', 'last', 'neither'] = 'first', opposite: bool = False, inplace: bool = True)
Filter out rows with duplicate names/IDs (index).
- Parameters:
keep ('first', 'last', or 'neither' (default='first')) – determines which of the duplicates to keep for each group of duplicates. ‘first’ will keep the first duplicate found for each group; ‘last’ will keep the last; and ‘neither’ will remove all of the values in the group.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- filter_fold_change_direction(direction: Literal['pos', 'neg'] = 'pos', opposite: bool = False, inplace: bool = True)
Filters out features according to the direction in which they changed between the two conditions.
- Parameters:
direction – ‘pos’ or ‘neg’. If ‘pos’, will keep only features that have positive log2foldchange. If ‘neg’, will keep only features that have negative log2foldchange.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current FoldChangeFilter object. If False, the function will return a new FoldChangeFilter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of FoldChangeFilter.
- Examples:
>>> from rnalysis import filtering >>> f = filtering.FoldChangeFilter('tests/test_files/fc_1.csv','numerator name','denominator name') >>> # keep only rows with a positive log2(fold change) value >>> f.filter_fold_change_direction('pos') Filtered 10 features, leaving 12 of the original 22 features. Filtered inplace.
>>> f = filtering.FoldChangeFilter('tests/test_files/fc_1.csv','numerator name','denominator name') >>> # keep only rows with a negative log2(fold change) value >>> f.filter_fold_change_direction('neg') Filtered 14 features, leaving 8 of the original 22 features. Filtered inplace.
>>> f = filtering.FoldChangeFilter('tests/test_files/fc_1.csv','numerator name','denominator name') >>> # keep only rows with a non-positive log2(fold change) value >>> f.filter_fold_change_direction('pos', opposite=True) Filtered 12 features, leaving 10 of the original 22 features. Filtered inplace.
- filter_missing_values(opposite: bool = False, inplace: bool = True)
Remove all rows with missing values.
- Parameters:
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- filter_percentile(percentile: Fraction, column: ColumnName, interpolate: 'nearest', 'higher', 'lower', 'midpoint', 'linear' = 'linear', opposite: bool = False, inplace: bool = True)
Removes all entries above the specified percentile in the specified column. For example, if the column were ‘pvalue’ and the percentile was 0.5, then all features whose pvalue is above the median pvalue will be filtered out.
- Parameters:
percentile (float between 0 and 1) – The percentile that all features above it will be filtered out.
column (str) – Name of the DataFrame column according to which the filtering will be performed.
interpolate ('nearest', 'higher', 'lower', 'midpoint' or 'linear' (default='linear')) – interpolation method to use when the desired quantile lies between two data points.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and filtered instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # keep only the rows whose value in the column 'log2FoldChange' is below the 75th percentile >>> d.filter_percentile(0.75,'log2FoldChange') Filtered 7 features, leaving 21 of the original 28 features. Filtered inplace.
>>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> # keep only the rows vulse value in the column 'log2FoldChange' is above the 25th percentile >>> d.filter_percentile(0.25,'log2FoldChange',opposite=True) Filtered 7 features, leaving 21 of the original 28 features. Filtered inplace.
- filter_top_n(by: ColumnNames, n: PositiveInt = 100, ascending: bool | List[bool] = True, na_position: str = 'last', opposite: bool = False, inplace: bool = True)
Sort the rows by the values of specified column or columns, then keep only the top ‘n’ rows.
- Parameters:
by (name of column/columns (str/List[str])) – Names of the column or columns to sort and then filter by.
n (int) – How many features to keep in the Filter object.
ascending (bool or list of bools (default=True)) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of Filter.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only the 10 rows with the highest values in the columns 'cond1' >>> counts.filter_top_n(by='cond1',n=10, ascending=False) Filtered 12 features, leaving 10 of the original 22 features. Filtered inplace.
>>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # keep only the 10 rows which have the lowest values in the columns 'cond1' >>> # and then the highest values in the column 'cond2' >>> counts.filter_top_n(by=['cond1','cond2'],n=10, ascending=[True,False]) Filtered 12 features, leaving 10 of the original 22 features. Filtered inplace.
- find_paralogs_ensembl(organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True)
Find paralogs within the same species using the Ensembl database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
- Returns:
DataFrame describing all discovered paralog mappings.
- find_paralogs_panther(organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto')
Find paralogs within the same species using the PantherDB database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- Returns:
DataFrame describing all discovered paralog mappings.
- fname
- head(n: PositiveInt = 5) DataFrame
Return the first n rows of the Filter object. See polars.DataFrame.head documentation.
- Parameters:
n (positive int, default 5) – Number of rows to show.
- Returns:
returns the first n rows of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> d.head() shape: (5, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬───────────┬─────────────┬─────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪═══════════╪═════════════╪═════════════╡ │ WBGene00000002 ┆ 6820.755327 ┆ 7.567762 ┆ 0.142257 ┆ 53.197692 ┆ 0.0 ┆ 0.0 │ │ WBGene00000003 ┆ 3049.62567 ┆ 9.138071 ┆ 0.245989 ┆ 37.148359 ┆ 4.6600e-302 ┆ 4.2800e-298 │ │ WBGene00000004 ┆ 1432.911791 ┆ 8.111737 ┆ 0.246801 ┆ 32.867521 ┆ 6.4000e-237 ┆ 3.9200e-233 │ │ WBGene00000005 ┆ 4028.154186 ┆ 6.534112 ┆ 0.202467 ┆ 32.272543 ┆ 1.7000e-228 ┆ 7.8000e-225 │ │ WBGene00000006 ┆ 1230.58524 ┆ 7.157428 ┆ 0.227948 ┆ 31.399325 ┆ 2.0700e-216 ┆ 7.5900e-213 │ └────────────────┴─────────────┴────────────────┴──────────┴───────────┴─────────────┴─────────────┘
>>> d.head(3) # return only the first 3 rows shape: (3, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬───────────┬─────────────┬─────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪═══════════╪═════════════╪═════════════╡ │ WBGene00000002 ┆ 6820.755327 ┆ 7.567762 ┆ 0.142257 ┆ 53.197692 ┆ 0.0 ┆ 0.0 │ │ WBGene00000003 ┆ 3049.62567 ┆ 9.138071 ┆ 0.245989 ┆ 37.148359 ┆ 4.6600e-302 ┆ 4.2800e-298 │ │ WBGene00000004 ┆ 1432.911791 ┆ 8.111737 ┆ 0.246801 ┆ 32.867521 ┆ 6.4000e-237 ┆ 3.9200e-233 │ └────────────────┴─────────────┴────────────────┴──────────┴───────────┴─────────────┴─────────────┘
- property index_set: set
Returns all of the features in the current DataFrame (which were not removed by previously used filter methods) as a set. if any duplicate features exist in the filter object (same WBGene appears more than once), the corresponding WBGene index will appear in the returned set ONLY ONCE.
- Returns:
A set of WBGene names.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> myset = counts.index_set >>> print(myset) {'WBGene00044022', 'WBGene00077504', 'WBGene00007079', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00077503', 'WBGene00007078', 'WBGene00007064', 'WBGene00077502', 'WBGene00044951', 'WBGene00007077', 'WBGene00007066', 'WBGene00007076', 'WBGene00014997', 'WBGene00043990', 'WBGene00007074', 'WBGene00043987', 'WBGene00007071', 'WBGene00043989', 'WBGene00043988', 'WBGene00007075'}
- property index_string: str
Returns a string of all feature indices in the current DataFrame, sorted by their current order in the FIlter object, and separated by newline.
- This includes all of the feature indices which were not filtered out by previously-used filter methods.
if any duplicate features exist in the filter object (same index appears more than once),
the corresponding index will appear in the returned string ONLY ONCE.
- Returns:
A string of WBGene indices separated by newlines (\n).
For example, “WBGene00000001\nWBGene00000003\nWBGene12345678”.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.sort(by='cond1',ascending=False) >>> mystring = counts.index_string >>> print(mystring) WBGene00007075 WBGene00043988 WBGene00043990 WBGene00007079 WBGene00007076 WBGene00043989 WBGene00007063 WBGene00007077 WBGene00007078 WBGene00007071 WBGene00007064 WBGene00007066 WBGene00007074 WBGene00043987 WBGene00007067 WBGene00014997 WBGene00044022 WBGene00077503 WBGene00077504 WBGene00077502 WBGene00007069 WBGene00044951
- intersection(*others: Filter | set, return_type: Literal['set', 'str'] = 'set', inplace: bool = False)
Keep only the features that exist in ALL of the given Filter objects/sets. Can be done either inplace on the first Filter object, or return a set/string of features.
- Parameters:
others (Filter or set objects.) – Objects to calculate intersection with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the intersecting features. If ‘str’, returns a string of the intersecting features, delimited by a comma.
inplace (bool (default=False)) – If True, the function will be applied in-place to the current Filter object. If False (default), the function will return a set/str that contains the intersecting indices.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that intersect between the given Filter objects/sets.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> # calculate intersection and return a set >>> d.intersection(a_set) {'WBGene00000002', 'WBGene00000003'}
# calculate intersection and filter in-place >>> d.intersection(a_set, inplace=True) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
- majority_vote_intersection(*others: Filter | set, majority_threshold: float = 0.5, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the features that appear in at least (majority_threhold * 100)% of the given Filter objects/sets. Majority-vote intersection with majority_threshold=0 is equivalent to Union. Majority-vote intersection with majority_threshold=1 is equivalent to Intersection.
- Parameters:
others (Filter or set objects.) – Objects to calculate intersection with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the intersecting WBGene indices. If ‘str’, returns a string of the intersecting indices, delimited by a comma.
majority_threshold (float (default=0.5)) – The threshold that determines what counts as majority. Features will be returned only if they appear in at least (majority_threshold * 100)% of the given Filter objects/sets.
- Return type:
set or str
- Returns:
If inplace=False, returns a set/string of the features that uphold majority vote intersection between two given Filter objects/sets.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> a_set = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> b_set = {'WBGene00000002','WBGene00000004'} >>> # calculate majority-vote intersection and return a set >>> d.majority_vote_intersection(a_set, b_set, majority_threshold=2/3) {'WBGene00000002', 'WBGene00000003', 'WBGene00000004'}
- map_orthologs_ensembl(map_to_organism: str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'], map_from_organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the Ensembl database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_orthoinspector(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the OrthoInspector database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_panther(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_least_diverged: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PantherDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_least_diverged (bool (default=True)) – if True (default), RNAlysis will only fetch ortholog mappings that were flagged as a ‘least diverged ortholog’ on the PantherDB database. You can read more about this flag on the PantherDB website: https://www.pantherdb.org/genes/
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_phylomedb(map_to_organism: str | int | Literal, map_from_organism: Literal['auto'] | str | int | Literal = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', consistency_score_threshold: Fraction = 0.5, filter_consistency_score: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PhylomeDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- param map_to_organism:
organism name or NCBI taxon ID of the target species for ortholog mapping.
- type map_to_organism:
str or int
- param map_from_organism:
organism name or NCBI taxon ID of the input genes’ source species.
- type map_from_organism:
str or int
- param gene_id_type:
the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- type gene_id_type:
str or ‘auto’ (default=’auto’)
- ` :param consistency_score_threshold: the minimum consistency score required for an ortholog mapping to be considered valid. Consistency scores are calculated by PhylomeDB and represent the confidence of the ortholog mapping. setting consistency_score_threshold to 0 will keep all mappings. You can read more about PhylomeDB consistency score on the PhylomeDB website: orthology.phylomedb.org/help
- type consistency_score_threshold:
float between 0 and 1 (default=0.5)
- param filter_consistency_score:
if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest consistency score.
- type filter_consistency_score:
bool (default=True)
- param non_unique_mode:
How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
- type non_unique_mode:
‘first’, ‘last’, ‘random’, or ‘none’ (default=’first’)
- param remove_unmapped_genes:
if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
- type remove_unmapped_genes:
bool (default=False)
- type inplace:
bool (default=True)
- param inplace:
If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- return:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- number_filters(column: ColumnName, operator: Literal['greater than', 'equals', 'lesser than', 'abs greater than'], value: float, opposite: bool = False, inplace: bool = True)
Applay a number filter (greater than, equal, lesser than) on a particular column in the Filter object.
- Parameters:
column (str) – name of the column to filter by
operator (str: 'gt' / 'greater than' / '>', 'eq' / 'equals' / '=', 'lt' / 'lesser than' / '<') – the operator to filter the column by (greater than, equal or lesser than)
value (float) – the value to filter by
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','gt',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
>>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','greater than',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
>>> filt = filtering.Filter('tests/test_files/test_deseq.csv') >>> #keep only rows that have a value greater than 5900 in the column 'baseMean'. >>> filt.number_filters('baseMean','>',5900) Filtered 26 features, leaving 2 of the original 28 features. Filtered inplace.
- numerator
name of the numerator
- print_features()
Print the feature indices in the Filter object, sorted by their current order in the FIlter object, and separated by newline.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.sort(by='cond1',ascending=False) >>> counts.print_features() WBGene00007075 WBGene00043988 WBGene00043990 WBGene00007079 WBGene00007076 WBGene00043989 WBGene00007063 WBGene00007077 WBGene00007078 WBGene00007071 WBGene00007064 WBGene00007066 WBGene00007074 WBGene00043987 WBGene00007067 WBGene00014997 WBGene00044022 WBGene00077503 WBGene00077504 WBGene00077502 WBGene00007069 WBGene00044951
- randomization_test(ref, alpha: Fraction = 0.05, reps: PositiveInt = 10000, save_csv: bool = False, fname: str | None = None, random_seed: int | None = None) DataFrame
Perform a randomization test to examine whether the fold change of a group of specific genomic features is significantly different than the fold change of a background set of genomic features.
- Parameters:
ref (FoldChangeFilter) – A reference FoldChangeFilter object which contains the fold change for every reference gene. Will be used to calculate the expected score and to perform randomizations.
alpha (float between 0 and 1) – Indicates the threshold for significance (alpha).
reps (int larger than 0) – How many repetitions to run the randomization for. 10,000 is the default. Recommended 10,000 or higher.
save_csv (bool, default False) – If True, will save the results to a .csv file, under the name specified in ‘fname’.
fname (str or pathlib.Path) – The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt.
- Return type:
pl.DataFrame
- Returns:
A Dataframe with the number of given genes, the observed fold change for the given group of genes, the expected fold change for a group of genes of that size and the p value for the comparison.
- Examples:
>>> from rnalysis import filtering >>> f = filtering.FoldChangeFilter('tests/test_files/fc_1.csv' , 'numerator' , 'denominator') >>> f_background = f.filter_biotype_from_ref_table('protein_coding', ref='tests/test_files/biotype_ref_table_for_tests.csv', inplace=False) #keep only protein-coding genes as reference Filtered 9 features, leaving 13 of the original 22 features. Filtering result saved to new object. >>> f_test = f_background.filter_by_attribute('attribute1', ref='tests/test_files/attr_ref_table_for_examples.csv', inplace=False) Filtered 6 features, leaving 7 of the original 13 features. Filtering result saved to new object. >>> rand_test_res = f_test.randomization_test(f_background) Calculating... shape: (1, 5) ┌────────────┬──────────────────────┬──────────────────────┬──────────┬─────────────┐ │ group size ┆ observed fold change ┆ expected fold change ┆ pval ┆ significant │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ f64 ┆ f64 ┆ f64 ┆ f64 ┆ bool │ ╞════════════╪══════════════════════╪══════════════════════╪══════════╪═════════════╡ │ 7.0 ┆ 2.806873 ┆ 2.510859 ┆ 0.354265 ┆ false │ └────────────┴──────────────────────┴──────────────────────┴──────────┴─────────────┘
- save_csv(alt_filename: None | str | Path = None)
Saves the current filtered data to a .csv file.
- Parameters:
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- save_parquet(alt_filename: None | str | Path = None)
Saves the current filtered data to a .parquet file.
- Parameters:
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- save_table(suffix: Literal['.csv', '.tsv', '.parquet'] = '.csv', alt_filename: None | str | Path = None)
Save the current filtered data table.
- Parameters:
suffix ('.csv', '.tsv', or '.parquet' (default='.csv')) – the file suffix
alt_filename (str, pathlib.Path, or None (default)) – If None, file name will be generated automatically according to the filtering methods used. If it’s a string, it will be used as the name of the saved file. Example input: ‘myfilename’
- sort(by: str | List[str], ascending: bool | List[bool] = True, na_position: str = 'last', inplace: bool = True)
Sort the rows by the values of specified column or columns.
- Parameters:
by (str or list of str) – Names of the column or columns to sort by.
ascending (bool or list of bool (default=True)) – Sort ascending vs. descending. Specify list for multiple sort orders. If this is a list of bools, it must have the same length as ‘by’.
na_position ('first' or 'last', default 'last') – If ‘first’, puts NaNs at the beginning; if ‘last’, puts NaNs at the end.
inplace (bool (default=True)) – If True, perform operation in-place. Otherwise, returns a sorted copy of the Filter object without modifying the original.
- Returns:
None if inplace=True, a sorted Filter object otherwise.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> counts.head() cond1 cond2 cond3 cond4 WBGene00007063 633 451 365 388 WBGene00007064 60 57 20 23 WBGene00044951 0 0 0 1 WBGene00007066 55 266 46 39 WBGene00007067 15 13 1 0 >>> counts.sort(by='cond1',ascending=True) >>> counts.head() cond1 cond2 cond3 cond4 WBGene00044951 0 0 0 1 WBGene00077504 0 0 0 0 WBGene00007069 0 2 1 0 WBGene00077502 0 0 0 0 WBGene00077503 1 4 2 0
- split_by_attribute(attributes: str | List[str], ref: str | Path | Literal['predefined'] = 'predefined') tuple
Splits the features in the Filter object into multiple Filter objects, each corresponding to one of the specified Attribute Reference Table attributes. Each new Filter object will contain only features that belong to its Attribute Reference Table attribute.
- Parameters:
attributes (list of strings) – list of attribute names from the Attribute Reference Table to filter by.
ref – filename/path of the reference table to be used as reference.
- Return type:
Tuple[filtering.Filter]
- Returns:
A tuple of Filter objects, each containing only features that match one Attribute Reference Table attribute; the Filter objects are returned in the same order the attributes were given in.
- Examples:
>>> from rnalysis import filtering >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> attribute1,attribute2 = counts.split_by_attribute(['attribute1','attribute2'], ... ref='tests/test_files/attr_ref_table_for_examples.csv') Filtered 15 features, leaving 7 of the original 22 features. Filtering result saved to new object. Filtered 20 features, leaving 2 of the original 22 features. Filtering result saved to new object.
- split_by_percentile(percentile: Fraction, column: ColumnName, interpolate: 'nearest', 'higher', 'lower', 'midpoint', 'linear' = 'linear') tuple
Splits the features in the Filter object into two non-overlapping Filter objects: one containing features below the specified percentile in the specfieid column, and the other containing features about the specified percentile in the specified column.
- Parameters:
percentile (float between 0 and 1) – The percentile that all features above it will be filtered out.
column (str) – Name of the DataFrame column according to which the filtering will be performed.
- Return type:
Tuple[filtering.Filter, filtering.Filter]
- Returns:
a tuple of two Filter objects: the first contains all of the features below the specified percentile, and the second contains all of the features above and equal to the specified percentile.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> below, above = d.split_by_percentile(0.75,'log2FoldChange') Filtered 7 features, leaving 21 of the original 28 features. Filtering result saved to new object. Filtered 21 features, leaving 7 of the original 28 features. Filtering result saved to new object.
- split_fold_change_direction() tuple
Splits the features in the FoldChangeFilter object into two non-overlapping FoldChangeFilter objects, based on the direction of their log2(fold change). The first object will contain only features with a positive log2(fold change), the second object will contain only features with a negative log2(fold change). Features with log2(fold change) = 0 will be ignored.
- Return type:
Tuple[filtering.FoldChangeFilter, filtering.FoldChangeFilter]
- Returns:
a tuple containing two FoldChangeFilter objects: the first has only features with positive log2 fold change, and the other has only features with negative log2 fold change.
- Examples:
>>> from rnalysis import filtering >>> f = filtering.FoldChangeFilter('tests/test_files/fc_1.csv','numerator name','denominator name') >>> pos_log2fc, neg_log2fc = f.split_fold_change_direction() Filtered 10 features, leaving 12 of the original 22 features. Filtering result saved to new object. Filtered 14 features, leaving 8 of the original 22 features. Filtering result saved to new object.
- symmetric_difference(other: Filter | set, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the WBGene indices that exist either in the first Filter object/set OR the second, but NOT in both (set symmetric difference).
- Parameters:
other (Filter or set.) – a second Filter object/set to calculate symmetric difference with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the features that exist in exactly one Filter object. If ‘str’, returns a string of the features that exist in exactly one Filter object, delimited by a comma.
- Return type:
set or str
- Returns:
a set/string of the features that that exist t in exactly one Filter. (set symmetric difference).
- Examples:
>>> from rnalysis import filtering >>> d = filtering.DESeqFilter("tests/test_files/test_deseq.csv") >>> counts = filtering.CountFilter('tests/test_files/counted.csv') >>> # calculate difference and return a set >>> d.symmetric_difference(counts) {'WBGene00000017', 'WBGene00077504', 'WBGene00000024', 'WBGene00000010', 'WBGene00000020', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00007078', 'WBGene00000029', 'WBGene00000006', 'WBGene00007064', 'WBGene00000019', 'WBGene00000004', 'WBGene00007066', 'WBGene00014997', 'WBGene00000023', 'WBGene00007074', 'WBGene00000025', 'WBGene00043989', 'WBGene00043988', 'WBGene00000014', 'WBGene00000027', 'WBGene00000021', 'WBGene00044022', 'WBGene00007079', 'WBGene00000012', 'WBGene00000005', 'WBGene00077503', 'WBGene00000026', 'WBGene00000003', 'WBGene00000002', 'WBGene00077502', 'WBGene00044951', 'WBGene00007077', 'WBGene00000007', 'WBGene00000008', 'WBGene00007076', 'WBGene00000013', 'WBGene00043990', 'WBGene00043987', 'WBGene00007071', 'WBGene00000011', 'WBGene00000015', 'WBGene00000018', 'WBGene00000016', 'WBGene00000028', 'WBGene00007075', 'WBGene00000022', 'WBGene00000009'}
- tail(n: PositiveInt = 5) DataFrame
Return the last n rows of the Filter object. See polars.DataFrame.tail documentation.
- Parameters:
n (positive int, default 5) – Number of rows to show.
- Return type:
pl.DataFrame
- Returns:
returns the last n rows of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> d.tail() shape: (5, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬────────────┬────────────┬────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪════════════╪════════════╪════════════╡ │ WBGene00000025 ┆ 2236.185837 ┆ 2.477374 ┆ 0.129606 ┆ 19.114626 ┆ 1.9100e-81 ┆ 1.4600e-78 │ │ WBGene00000026 ┆ 343.648987 ┆ -4.037191 ┆ 0.219781 ┆ -18.369115 ┆ 2.3200e-75 ┆ 1.7000e-72 │ │ WBGene00000027 ┆ 175.142856 ┆ 6.352044 ┆ 0.347777 ┆ 18.264706 ┆ 1.5800e-74 ┆ 1.1200e-71 │ │ WBGene00000028 ┆ 219.1632 ┆ 3.913657 ┆ 0.217802 ┆ 17.968851 ┆ 3.4200e-72 ┆ 2.3200e-69 │ │ WBGene00000029 ┆ 1066.242402 ┆ -2.811281 ┆ 0.158284 ┆ -17.761002 ┆ 1.4200e-70 ┆ 9.2900e-68 │ └────────────────┴─────────────┴────────────────┴──────────┴────────────┴────────────┴────────────┘
>>> d.tail(8) # returns the last 8 rows shape: (8, 7) ┌────────────────┬─────────────┬────────────────┬──────────┬────────────┬────────────┬────────────┐ │ ┆ baseMean ┆ log2FoldChange ┆ lfcSE ┆ stat ┆ pvalue ┆ padj │ │ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- ┆ --- │ │ str ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 ┆ f64 │ ╞════════════════╪═════════════╪════════════════╪══════════╪════════════╪════════════╪════════════╡ │ WBGene00000022 ┆ 365.813048 ┆ 6.101303 ┆ 0.291484 ┆ 20.931891 ┆ 2.7400e-97 ┆ 2.4000e-94 │ │ WBGene00000023 ┆ 3168.566714 ┆ 3.906719 ┆ 0.190439 ┆ 20.514331 ┆ 1.6000e-93 ┆ 1.3400e-90 │ │ WBGene00000024 ┆ 221.925724 ┆ 4.801676 ┆ 0.246313 ┆ 19.494189 ┆ 1.2300e-84 ┆ 9.8200e-82 │ │ WBGene00000025 ┆ 2236.185837 ┆ 2.477374 ┆ 0.129606 ┆ 19.114626 ┆ 1.9100e-81 ┆ 1.4600e-78 │ │ WBGene00000026 ┆ 343.648987 ┆ -4.037191 ┆ 0.219781 ┆ -18.369115 ┆ 2.3200e-75 ┆ 1.7000e-72 │ │ WBGene00000027 ┆ 175.142856 ┆ 6.352044 ┆ 0.347777 ┆ 18.264706 ┆ 1.5800e-74 ┆ 1.1200e-71 │ │ WBGene00000028 ┆ 219.1632 ┆ 3.913657 ┆ 0.217802 ┆ 17.968851 ┆ 3.4200e-72 ┆ 2.3200e-69 │ │ WBGene00000029 ┆ 1066.242402 ┆ -2.811281 ┆ 0.158284 ┆ -17.761002 ┆ 1.4200e-70 ┆ 9.2900e-68 │ └────────────────┴─────────────┴────────────────┴──────────┴────────────┴────────────┴────────────┘
- text_filters(column: ColumnName, operator: Literal['equals', 'contains', 'starts with', 'ends with'], value: str, opposite: bool = False, inplace: bool = True)
Applay a text filter (equals, contains, starts with, ends with) on a particular column in the Filter object.
- Parameters:
column (str) – name of the column to filter by
operator (str: 'eq' / 'equals' / '=', 'ct' / 'contains' / 'in', 'sw' / 'starts with', 'ew' / 'ends with') – the operator to filter the column by (equals, contains, starts with, ends with)
value (number (int or float)) – the value to filter by
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/text_filters.csv') >>> # keep only rows that have a value that starts with 'AC3' in the column 'name'. >>> filt.text_filters('name','sw','AC3') Filtered 17 features, leaving 5 of the original 22 features. Filtered inplace.
- transform(function: Literal['Box-Cox', 'log2', 'log10', 'ln', 'Standardize'] | Callable, columns: ColumnNames | Literal['all'] = 'all', inplace: bool = True, **function_kwargs)
Transform the values in the Filter object with the specified function.
- Parameters:
function (Callable or str ('logx' for base-x log of the data + 1, 'box-cox' for Box-Cox transform of the data + 1, 'standardize' for standardization)) – The function or function name to be applied.
columns (str, list of str, or 'all' (default='all')) – The columns to which the transform should be applied.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
function_kwargs – Any additional keyworded arguments taken by the supplied function.
- Returns:
If ‘inplace’ is False, returns a new instance of the Filter object.
- Examples:
>>> from rnalysis import filtering >>> filt = filtering.Filter('tests/test_files/counted.csv') >>> filt_log10 = filt.transform('log10', inplace=False) Transformed 22 features. Transformation result saved to new object. >>> filt.transform(lambda x: x+1, columns=['cond1','cond4']) Transformed 22 features. Transformed inplace.
- translate_gene_ids(translate_to: str | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'], translate_from: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', remove_unmapped_genes: bool = False, inplace: bool = True)
Translates gene names/IDs from one type to another. Mapping is done using the UniProtKB Gene ID Mapping service. You can choose to optionally drop from the table all rows that failed to be translated.
- Parameters:
translate_to (str) – the gene ID type to translate gene names/IDs to. For example: UniProtKB, Ensembl, Wormbase.
translate_from (str or 'auto' (default='auto')) – the gene ID type to translate gene names/IDs from. For example: UniProtKB, Ensembl, Wormbase. If translate_from=’auto’, RNAlysis will attempt to automatically determine the gene ID type of the features in the table.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be translated will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If inplace is False, returns a new and translated instance of the Filter object.
- union(*others: Filter | set, return_type: Literal['set', 'str'] = 'set')
Returns a set/string of the union of features between multiple Filter objects/sets (the features that exist in at least one of the Filter objects/sets).
- Parameters:
others (Filter or set objects.) – Objects to calculate union with.
return_type ('set' or 'str' (default='set')) – If ‘set’, returns a set of the union features. If ‘str’, returns a string of the union WBGene indices, delimited by a comma.
- Return type:
set or str
- Returns:
a set/string of the WBGene indices that exist in at least one of the Filter objects.
- Examples:
>>> from rnalysis import filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> counts = filtering.Filter('tests/test_files/counted.csv') >>> # calculate union and return a set >>> d.union(counts) {'WBGene00000017', 'WBGene00000021', 'WBGene00044022', 'WBGene00077504', 'WBGene00000012', 'WBGene00000024', 'WBGene00007079', 'WBGene00000010', 'WBGene00000020', 'WBGene00000005', 'WBGene00007069', 'WBGene00007063', 'WBGene00007067', 'WBGene00077503', 'WBGene00007078', 'WBGene00000026', 'WBGene00000029', 'WBGene00000002', 'WBGene00000003', 'WBGene00000006', 'WBGene00007064', 'WBGene00077502', 'WBGene00044951', 'WBGene00000007', 'WBGene00000008', 'WBGene00000019', 'WBGene00007077', 'WBGene00000004', 'WBGene00007066', 'WBGene00007076', 'WBGene00000013', 'WBGene00014997', 'WBGene00000023', 'WBGene00043990', 'WBGene00007074', 'WBGene00000025', 'WBGene00000011', 'WBGene00043987', 'WBGene00007071', 'WBGene00000015', 'WBGene00000018', 'WBGene00043989', 'WBGene00043988', 'WBGene00000014', 'WBGene00000016', 'WBGene00000027', 'WBGene00000028', 'WBGene00007075', 'WBGene00000022', 'WBGene00000009'}
- class rnalysis.filtering.Pipeline(filter_type: str | ~rnalysis.filtering.Filter | ~rnalysis.filtering.CountFilter | ~rnalysis.filtering.DESeqFilter | ~rnalysis.filtering.FoldChangeFilter = <class 'rnalysis.filtering.Filter'>)
Bases:
GenericPipelineA collection of functions to be applied sequentially to Filter objects.
Attributes
- functions: list
A list of the functions in the Pipeline.
- params: list
A list of the parameters of the functions in the Pipeline.
- filter_type: Filter object
The type of Filter objects to which the Pipeline can be applied
- _apply_filter_norm_sort(func: LambdaType, filter_object: Filter | CountFilter | DESeqFilter | FoldChangeFilter, args: tuple, kwargs: dict, inplace: bool, other_outputs: dict, other_cnt)
Apply a filtering/normalizing/sorting function.
- Parameters:
func (function) – function to apply
filter_object (Filter, CountFilter, DESeqFilter, or FoldChangeFilter.) – Filter object to apply function to
args (tuple) – arguments for the function
kwargs (dict) – keyworded arguments for the function
inplace (bool) – if True, function will be applied inplace.
- Returns:
Filter object to which the function was applied.
- _apply_other(func: LambdaType, filter_object: Filter | CountFilter | DESeqFilter | FoldChangeFilter, args: tuple, kwargs: dict, other_outputs: dict, other_cnt: dict, recursive_call: bool = False)
Apply a non filtering/splitting/normalizing/sorting function.
- Parameters:
func (function) – function to apply
filter_object (Filter, CountFilter, DESeqFilter, or FoldChangeFilter.) – Filter object to apply function to
args (tuple) – arguments for the function
kwargs (dict) – keyworded arguments for the function
other_outputs (dict) – dictionary with additional function outputs
other_cnt (dict) – counter for how many times each function was already called
- Returns:
Filter object to which the function was applied.
- _apply_split(func: LambdaType, filter_object: Filter | CountFilter | DESeqFilter | FoldChangeFilter, args: tuple, kwargs: dict, other_outputs: dict, other_cnt: dict)
Apply a splitting function.
- Parameters:
func (function) – function to apply
filter_object (Filter, CountFilter, DESeqFilter, or FoldChangeFilter.) – Filter object to apply function to
args (tuple) – arguments for the function
kwargs (dict) – keyworded arguments for the function
other_outputs (dict) – dictionary with additional function outputs
other_cnt (dict) – counter for how many times each function was already called
- Returns:
Filter object to which the function was applied.
- _func_signature(func: LambdaType, args: tuple, kwargs: dict)
Returns a string functions signature for the given function and arguments.
- Parameters:
func (function) – the function or method to generate signature for
args (tuple) – arguments given for the function
kwargs (dict) – keyworded arguments given for the function
- Returns:
function signature string
- Return type:
str
- static _param_string(args: tuple, kwargs: dict)
Returns a formatted string of the given arguments and keyworded arguments.
- Parameters:
args (tuple) – arguments to format as string
kwargs (dict) – keyworded arguments to format as string
- Returns:
a formatted string of arguments and keyworded argumentss
- Return type:
str
- _readable_func_signature(func: LambdaType, args: tuple, kwargs: dict)
Returns a human-readable string functions signature for the given function and arguments.
- Parameters:
func (function) – the function or method to generate signature for
args (tuple) – arguments given for the function
kwargs (dict) – keyworded arguments given for the function
- Returns:
function signature string
- Return type:
str
- add_function(func: LambdaType | str, *args, **kwargs)
Add a function to the pipeline. Arguments can be stated with or without the correspoding keywords. For example: Pipeline.add_function(‘sort’, ‘columnName’, ascending=False, na_position=’first’). Pipelines support virtually all functions in the filtering module, including filtering functions, normalization functions, splitting functions and plotting functions. Do not include the ‘inplace’ argument when adding functions to a pipeline; instead, include it when applying the pipeline to a Filter object using Pipeline.apply_to(). :param func: function to be added :type func: function or name of function from the filtering module :param args: unkeyworded arguments for the added function in their natural order. For example: 0.1, True :param kwargs: keyworded arguments for the added function. For example: opposite=True
- Examples:
>>> from rnalysis import filtering >>> pipe = filtering.Pipeline() >>> pipe.add_function(filtering.Filter.filter_missing_values) Added function 'Filter.filter_missing_values()' to the pipeline. >>> pipe.add_function('number_filters', 'col1', 'greater than', value=5, opposite=True) Added function 'Filter.number_filters('col1', 'greater than', value=5, opposite=True)' to the pipeline.
- apply_to(filter_object: Filter | CountFilter | DESeqFilter | FoldChangeFilter, inplace: bool = True) Filter | Tuple[Filter, dict] | Tuple[Tuple[Filter], dict] | dict | None
Sequentially apply all functions in the Pipeline to a given Filter object.
- Parameters:
filter_object (Filter, CountFilter, DESeqFilter, or FoldChangeFilter) – filter object to apply the Pipeline to. Type of filter_object must be identical to Pipeline.filter_type.
inplace (bool (default=True)) – Determines whether to apply operations in-place or not.
- Returns:
If inplace=False, a Filter object/tuple of Filter objects will be returned. If the functions in the Pipeline return any additional outputs, they will also be returned in a dictionary. Otherwise, nothing will be returned.
- Return type:
Filter object, Tuple[Filter, dict], dict, or None
- Examples:
>>> from rnalysis import filtering >>> # create the pipeline >>> pipe = filtering.Pipeline('DESeqFilter') >>> pipe.add_function(filtering.DESeqFilter.filter_missing_values) Added function 'DESeqFilter.filter_missing_values()' to the pipeline. >>> pipe.add_function(filtering.DESeqFilter.filter_top_n, by='padj', n=3) Added function 'DESeqFilter.filter_top_n(by='padj', n=3)' to the pipeline. >>> pipe.add_function('sort', by='baseMean') Added function 'DESeqFilter.sort(by='baseMean')' to the pipeline. >>> # load the Filter object >>> d = filtering.DESeqFilter('tests/test_files/test_deseq_with_nan.csv') >>> # apply the Pipeline not-inplace >>> d_filtered = pipe.apply_to(d, inplace=False) Filtered 3 features, leaving 25 of the original 28 features. Filtering result saved to new object. Filtered 22 features, leaving 3 of the original 25 features. Filtering result saved to new object. Sorted 3 features. Sorting result saved to a new object. >>> # apply the Pipeline inplace >>> pipe.apply_to(d) Filtered 3 features, leaving 25 of the original 28 features. Filtered inplace. Filtered 22 features, leaving 3 of the original 25 features. Filtered inplace. Sorted 3 features. Sorted inplace.
- export_pipeline(filename: str | Path | None) None | str
Export a Pipeline to a Pipeline YAML file or YAML-like string.
- Parameters:
filename (str, pathlib.Path, or None) – filename to save the Pipeline YAML to, or None to return a YAML-like string instead.
- Returns:
if filename is None, returns the Pipeline YAML-like string.
- filter_type
type of filter objects to which the Pipeline can be applied
- functions
- classmethod import_pipeline(filename: str | Path) GenericPipeline
Import a Pipeline from a Pipeline YAML file or YAML-like string.
- Parameters:
filename (str or pathlib.Path) – name of the YAML file containing the Pipeline, or a YAML-like string.
- Returns:
the imported Pipeline
- Return type:
- params
- remove_last_function()
Removes from the Pipeline the last function that was added to it. Removal is in-place.
- Examples:
>>> from rnalysis import filtering >>> pipe = filtering.Pipeline() >>> pipe.add_function(filtering.Filter.filter_missing_values) Added function 'Filter.filter_missing_values()' to the pipeline. >>> pipe.remove_last_function() Removed function filter_missing_values with parameters [] from the pipeline.
- rnalysis.filtering.infer_table_type(fname: str | Path | None = None, df: DataFrame | Series | None = None) str
Infer the most likely table type of a data file, to use as the pre-selected default on the GUI’s “New table” screen. Detection is deliberately conservative: whenever the content is ambiguous it returns ‘Other table’ rather than risk a wrong (and silently limiting) classification that the user then has to undo. This only influences the pre-selected type – it never changes how a table is parsed or loaded – and it never raises: any error while reading or inspecting the data degrades to ‘Other table’.
For speed, when reading from a file only the header and a small sample of rows are read (the full table is not loaded on the UI thread). The differential-expression and fold-change signatures are decided from the column names alone; the count-matrix checks use the sampled rows. As a consequence, a table that only turns non-count-like (e.g. contains a negative value) below the sampled rows may still be pre-selected as a count matrix – an accepted, user-overridable limitation of the cheap read.
- Parameters:
fname (str, pathlib.Path, or None (default=None)) – path of the table file to inspect. Ignored if
dfis provided.df (polars.DataFrame, polars.Series, or None (default=None)) – an already-loaded polars DataFrame/Series to inspect instead of reading
fname.
- Returns:
one of ‘Count matrix’, ‘Differential expression’, ‘Fold change’, or ‘Other table’ (the keys used by the GUI table-type combo box).
- Return type:
str
rnalysis.enrichment module
This module can perform enrichment analyses on a given set of genomic features and visualize their intersections. These include gene ontology/tissue/phenotype enrichment, enrichment for user-defined attributes, set visualization ,etc. Results of enrichment analyses can be saved to .csv files.
- class rnalysis.enrichment.FeatureSet(gene_set: List[str] | Set[str] | Filter = None, set_name: str = '')
Bases:
setReceives a filtered gene set and the set’s name (optional) and preforms various enrichment analyses on them.
- _inplace(func, func_kwargs, inplace: bool, **update_kwargs)
Executes the user’s choice whether to filter in-place or create a new instance of the FeatureSet object.
- _set_ops(others: set | FeatureSet | Tuple[set | FeatureSet], op: LambdaType) set
Performs a given set operation on self and on another object (FeatureSet or set). :type others: FeatureSet or set :param others: Other object to perform set operation with. :type: op: Callable (set.union, set.intersection, set.difference or set.symmetric difference) :param op: The set operation to be performed. :return: A set resulting from the set operation.
- add()
Add an element to a set.
This has no effect if the element is already present.
- biotypes_from_gtf(gtf_path: str | Path, attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene') DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- biotypes_from_ref_table(ref: str | Path | Literal['predefined'] = 'predefined')
Returns a DataFrame describing the biotypes in the gene set and their count.
- Parameters:
ref (str or pathlib.Path (default='predefined')) – Path of the reference file used to determine biotype. Default is the path predefined in the settings file.
- Examples:
>>> from rnalysis import enrichment, filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> en = enrichment.FeatureSet(d) >>> en.biotypes_from_ref_table(ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 2) ┌────────────────┬───────┐ │ biotype ┆ count │ │ --- ┆ --- │ │ str ┆ u32 │ ╞════════════════╪═══════╡ │ protein_coding ┆ 26 │ │ pseudogene ┆ 1 │ │ unknown ┆ 1 │ └────────────────┴───────┘
- change_set_name(new_name: str)
Change the ‘set_name’ of a FeatureSet to a new name.
- Parameters:
new_name (str) – the new set name
- clear()
Remove all elements from this set.
- copy()
Return a shallow copy of a set.
- difference(*others: set | FeatureSet) FeatureSet
Calculates the set difference of the indices from multiple FeatureSet objects (the indices that appear in the first FeatureSet object but NOT in the other objects).
- Parameters:
others (FeatureSet, RankedSet or set) – The objects against which the current object will be compared.
- Returns:
a new FeatureSet with elements in this FeatureSet that are not in the other objects.
- Return type:
- Examples:
>>> from rnalysis import enrichment >>> en = enrichment.FeatureSet({'WBGene00000001','WBGene00000002','WBGene00000006'}, 'set name') >>> s = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> en2 = enrichment.FeatureSet({'WBGene00000004','WBGene00000001'}) >>> en.difference(s, en2) >>> print(en) FeatureSet: set name {'WBGene00000006'}
- difference_update()
Remove all elements of another set from this set.
- discard()
Remove an element from a set if it is a member.
Unlike set.remove(), the discard() method does not raise an exception when an element is missing from the set.
- filter_biotype_from_gtf(gtf_path: str | Path, biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
biotype (str or list of strings) – the biotypes which will not be filtered out.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- filter_biotype_from_ref_table(biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
- Parameters:
biotype (string or list of strings) – the biotypes which will not be filtered out.
ref – Name of the biotype reference file used to determine biotypes. Default is the path defined by the user in the settings.yaml file.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- filter_by_attribute(attributes: str | List[str], mode: Literal['union', 'intersection'] = 'union', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters features according to user-defined attributes from an Attribute Reference Table. When multiple attributes are given, filtering can be done in ‘union’ mode (where features that belong to at least one attribute are not filtered out), or in ‘intersection’ mode (where only features that belong to ALL attributes are not filtered out). To learn more about user-defined attributes and Attribute Reference Tables, read the user guide.
- Parameters:
attributes (string or list of strings, which are column titles in the user-defined Attribute Reference Table.) – attributes to filter by.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
ref (str or pathlib.Path (default='predefined')) – filename/path of the attribute reference table to be used as reference.
opposite (bool (default=False)) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- filter_by_go_annotations(go_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', propagate_annotations: bool = True, evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', opposite: bool = False, inplace: bool = True)
Filters genes according to GO annotations, keeping only genes that are annotated with a specific GO term. When multiple GO terms are given, filtering can be done in ‘union’ mode (where genes that belong to at least one GO term are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL GO terms are not filtered out).
- Parameters:
go_ids (str or list of str)
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :param propagate_annotations: determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/ :type propagate_annotations: ‘classic’, ‘elim’, ‘weight’, ‘all.m’, or ‘no’ (default=’elim’) :param evidence_types: only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or ‘any’ (default=’any’) :param excluded_evidence_types: annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type excluded_evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or None (default=None) :param databases: only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current FeatureSet object. If False, the function will return a new FeatureSet instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the FeatureSet object.
- filter_by_kegg_annotations(kegg_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', opposite: bool = False, inplace: bool = True)
Filters genes according to KEGG pathways, keeping only genes that belong to specific KEGG pathway. When multiple KEGG IDs are given, filtering can be done in ‘union’ mode (where genes that belong to at least one pathway are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL pathways are not filtered out).
- Parameters:
kegg_ids (str or list of str) – the KEGG pathway IDs according to which the table will be filtered. An example for a legal KEGG pathway ID would be ‘path:cel04020’ for the C. elegans calcium signaling pathway.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current FeatureSet object. If False, the function will return a new Filter instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the FeatureSet object.
- find_paralogs_ensembl(organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True)
Find paralogs within the same species using the Ensembl database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
- Returns:
DataFrame describing all discovered paralog mappings.
- find_paralogs_panther(organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto')
Find paralogs within the same species using the PantherDB database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- Returns:
DataFrame describing all discovered paralog mappings.
- gene_set
set of feature names/indices
- go_enrichment(background_genes: Set[str] | Filter | FeatureSet, organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', alpha: Fraction = 0.05, statistical_test: Literal['fisher', 'hypergeometric', 'randomization'] = 'fisher', propagate_annotations: Literal['classic', 'elim', 'weight', 'all.m', 'no'] = 'elim', aspects: Literal['any', 'biological_process', 'cellular_component', 'molecular_function'] | Iterable[Literal['biological_process', 'cellular_component', 'molecular_function']] = 'any', evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] | Literal['any'] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', exclude_unannotated_genes: bool = True, return_nonsignificant: bool = False, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', plot_ontology_graph: bool = True, ontology_graph_format: Literal['pdf', 'png', 'svg', 'none'] = 'none', randomization_reps: PositiveInt = 10000, random_seed: int | None = None, parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) DataFrame | Tuple[DataFrame, Figure]
Calculates enrichment and depletion of the FeatureSet for Gene Ontology (GO) terms against a background set. The GO terms and annotations are drawn via the GO Solr search engine GOlr, using the search terms defined by the user. The background set is determined by either the input variable ‘background_genes’, or by the input variable ‘biotype’ and a Biotype Reference Table. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment score = 0) appears with the smallest value in the scale.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID for which the function will fetch GO annotations.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the GOLR server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
alpha (float between 0 and 1 (default=0.05)) – Indicates the FDR threshold for significance.
statistical_test ('fisher', 'hypergeometric' or 'randomization' (default='fisher')) – determines the statistical test to be used for enrichment analysis. Note that some propagation methods support only some of the available statistical tests.
biotype (str specifying a specific biotype, list/set of strings each specifying a biotype, or 'all'. Default 'protein_coding'.) – determines the background genes by their biotype. Requires specifying a Biotype Reference Table. ‘all’ will include all genomic features in the reference table, ‘protein_coding’ will include only protein-coding genes from the reference table, etc. Cannot be specified together with ‘background_genes’.
background_genes (set of feature indices, filtering.Filter object, or enrichment.FeatureSet object) – a set of specific feature indices to be used as background genes.
biotype_ref_path (str or pathlib.Path (default='predefined')) – the path of the Biotype Reference Table. Will be used to generate background set if ‘biotype’ is specified.
propagate_annotations ('classic', 'elim', 'weight', 'all.m', or 'no' (default='elim')) – determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/
aspects (str, Iterable of str, 'biological_process', 'molecular_function', 'cellular_component', or 'any' (default='any')) – only annotations from the specified GO aspects will be included in the analysis. Legal aspects are ‘biological_process’ (P), ‘molecular_function’ (F), and ‘cellular_component’ (C).
evidence_types (str, Iterable of str, 'experimental', 'phylogenetic' ,'computational', 'author', 'curator', 'electronic', or 'any' (default='any')) – only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/
excluded_evidence_types (str, Iterable of str, 'experimental', 'phylogenetic' ,'computational', 'author', 'curator', 'electronic', or None (default=None)) – annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/
databases – only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website:
http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :param exclude_unannotated_genes: if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results. :type exclude_unannotated_genes: bool (deafult=True) :param return_nonsignificant: if True, the results DataFrame will include all tested GO terms - both significant and non-significant terms. If False (default), only significant GO terms will be returned. :type return_nonsignificant: bool (default=False) :type save_csv: bool, default False :param save_csv: If True, will save the results to a .csv file, under the name specified in ‘fname’. :type fname: str or pathlib.Path :param fname: The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt. :type return_fig: bool (default=False) :param return_fig: if True, returns a matplotlib Figure object in addition to the results DataFrame. :type plot_ontology_graph: bool (default=True) :param plot_ontology_graph: if True, will generate an ontology graph depicting the significant GO terms and their parent nodes. :type ontology_graph_format: ‘pdf’, ‘png’, ‘svg’, or ‘none’ (default=’none’) :param ontology_graph_format: if ontology_graph_format is not ‘none’, the ontology graph will additonally be generated in the specified file format. :type plot_horizontal: bool (default=True) :param plot_horizontal: if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot. :param show_expected: if True, the observed/expected values will be shown on the plot. :type show_expected: bool (default=False) :param plot_style: style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set. :type plot_style: ‘bar’ or ‘lollipop’ (default=’bar’) :type random_seed: non-negative integer (default=None) :type random_seed: if using a randomization test, determine the random seed used to initialize the pseudorandom generator for the randomization test. By default it is picked at random, but you can set it to a particular integer to get consistents results over multiple runs. If not using a randomization test, this parameter will not affect the analysis. :param randomization_reps: if using a randomization test, determine how many randomization repititions to run. Otherwise, this parameter will not affect the analysis. :type randomization_reps: int larger than 0 (default=10000) :type parallel_backend: Literal[PARALLEL_BACKENDS] (default=’loky’) :param parallel_backend: Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise. :rtype: pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure] :return: a polars DataFrame with GO terms in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
Example plot of go_enrichment(plot_ontology_graph=True)
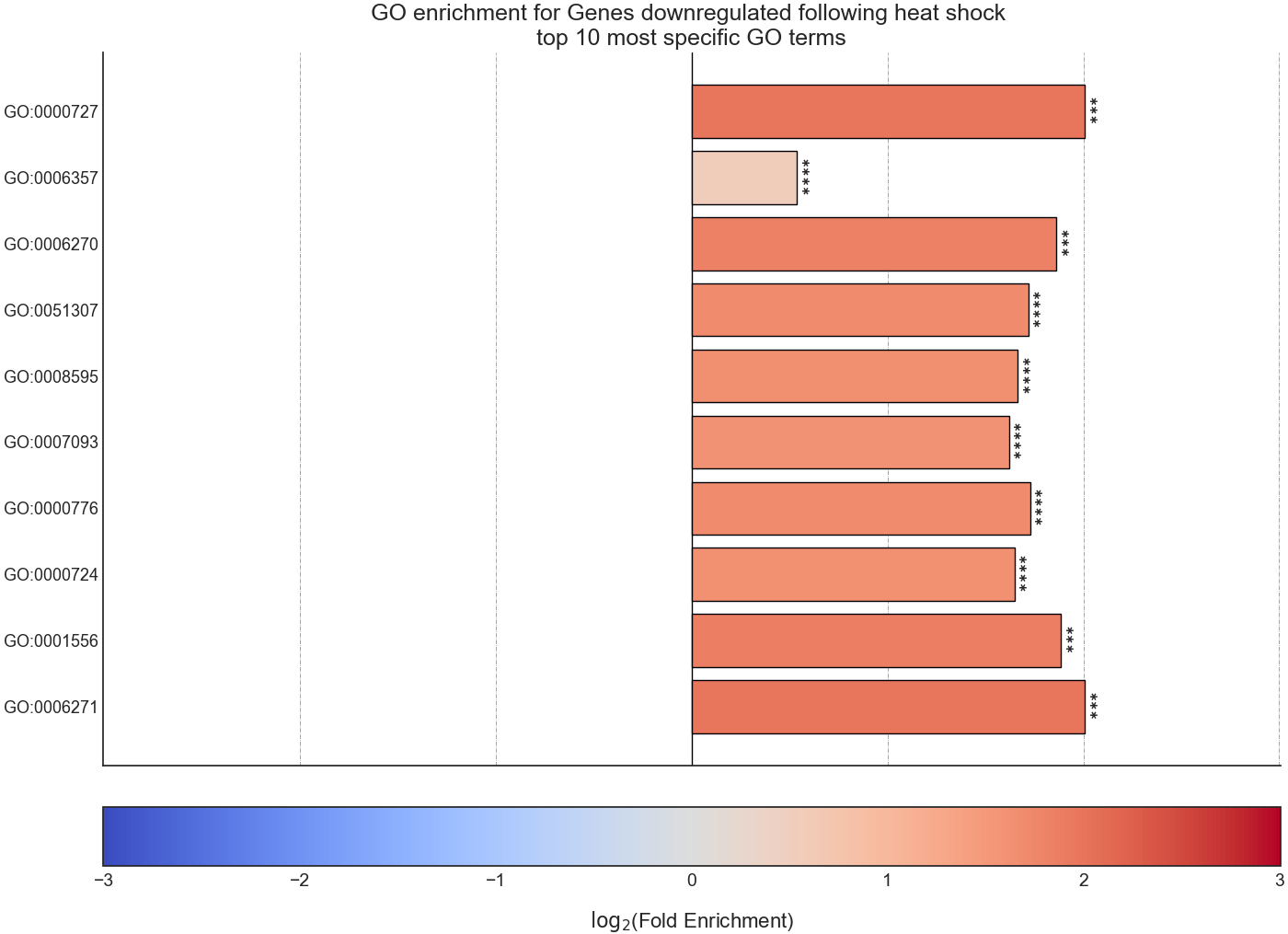
Example plot of go_enrichment()
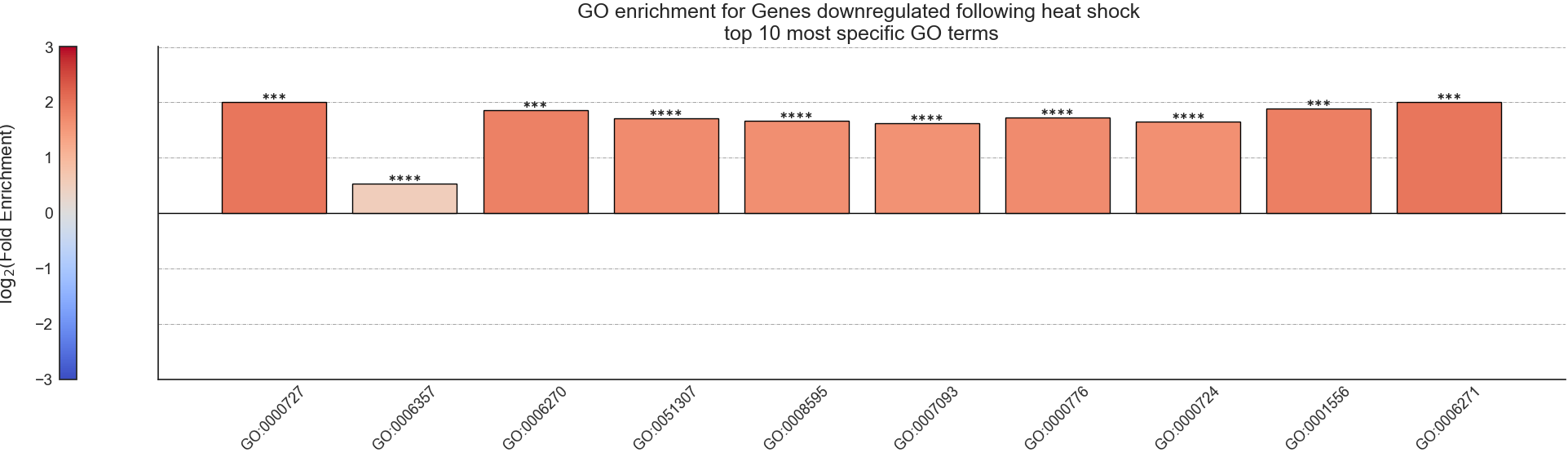
Example plot of go_enrichment(plot_horizontal = False)
- intersection(*others: set | FeatureSet) FeatureSet
Calculates the set intersection of the indices from multiple FeatureSet objects (the indices that exist in ALL of the FeatureSet objects).
- Parameters:
others (FeatureSet, RankedSet or set) – The objects against which the current object will be compared.
- Returns:
a new FeatureSet with elements common to this FeatureSet and all other objects.
- Return type:
- Examples:
>>> from rnalysis import enrichment >>> en = enrichment.FeatureSet({'WBGene00000001','WBGene00000002','WBGene00000006'}, 'set name') >>> s = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> en2 = enrichment.FeatureSet({'WBGene00000004','WBGene00000001'}) >>> en.intersection(s, en2) >>> print(en) FeatureSet: set name {'WBGene00000001'}
- intersection_update()
Update a set with the intersection of itself and another.
- isdisjoint()
Return True if two sets have a null intersection.
- issubset(other, /)
Test whether every element in the set is in other.
- issuperset(other, /)
Test whether every element in other is in the set.
- kegg_enrichment(background_genes: Set[str] | Filter | FeatureSet, organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', alpha: Fraction = 0.05, statistical_test: Literal['fisher', 'hypergeometric', 'randomization'] = 'fisher', exclude_unannotated_genes: bool = True, return_nonsignificant: bool = False, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', plot_pathway_graphs: bool = True, pathway_graphs_format: Literal['pdf', 'png', 'svg', 'none'] = 'none', randomization_reps: PositiveInt = 10000, random_seed: int | None = None, parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) DataFrame | Tuple[DataFrame, Figure]
Calculates enrichment and depletion of the FeatureSet for Kyoto Encyclopedia of Genes and Genomes (KEGG) curated pathways against a background set. The background set is determined by either the input variable ‘background_genes’, or by the input variable ‘biotype’ and a Biotype Reference Table. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment score = 0) appears with the smallest value in the scale.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID for which the function will fetch GO annotations.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
alpha (float between 0 and 1 (default=0.05)) – Indicates the FDR threshold for significance.
statistical_test ('fisher', 'hypergeometric' or 'randomization' (default='fisher')) – determines the statistical test to be used for enrichment analysis. Note that some propagation methods support only some of the available statistical tests.
background_genes (set of feature indices, filtering.Filter object, or enrichment.FeatureSet object) – a set of specific feature indices to be used as background genes.
exclude_unannotated_genes (bool (deafult=True)) – if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results.
return_nonsignificant (bool (default=False)) – if True, the results DataFrame will include all tested pathways - both significant and non-significant ones. If False (default), only significant pathways will be returned.
save_csv (bool, default False) – If True, will save the results to a .csv file, under the name specified in ‘fname’.
fname (str or pathlib.Path) – The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt.
return_fig (bool (default=False)) – if True, returns a matplotlib Figure object in addition to the results DataFrame.
plot_horizontal (bool (default=True)) – if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot.
show_expected (bool (default=False)) – if True, the observed/expected values will be shown on the plot.
plot_style ('bar' or 'lollipop' (default='bar')) – style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set.
plot_pathway_graphs (bool (default=True)) – if True, will generate pathway graphs depicting the significant KEGG pathways.
pathway_graphs_format ('pdf', 'png', 'svg', or None (default=None)) – if pathway_graphs_format is not ‘none’, the pathway graphs will additonally be generated in the specified file format.
randomization_reps (int larger than 0 (default=10000)) – if using a randomization test, determine how many randomization repititions to run. Otherwise, this parameter will not affect the analysis.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Return type:
pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure]
- Returns:
a polars DataFrame with the indicated pathway names in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
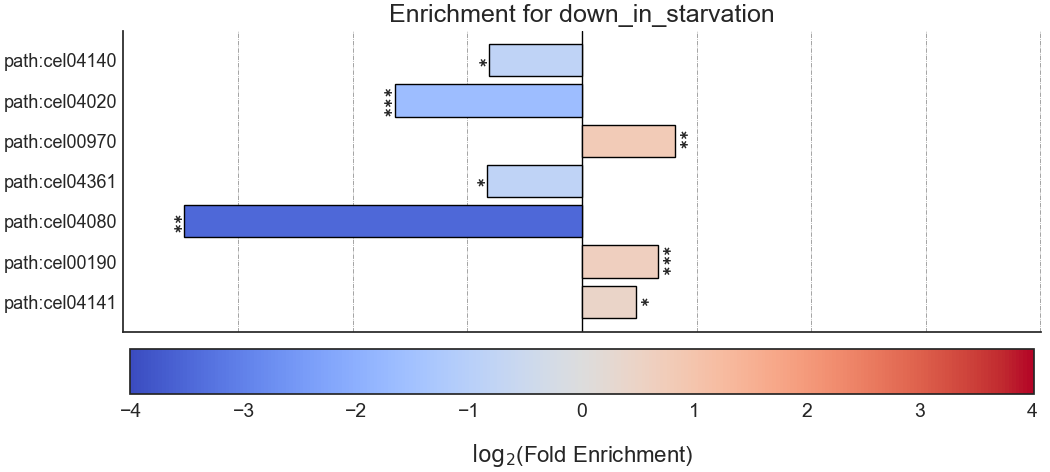
Example plot of kegg_enrichment()
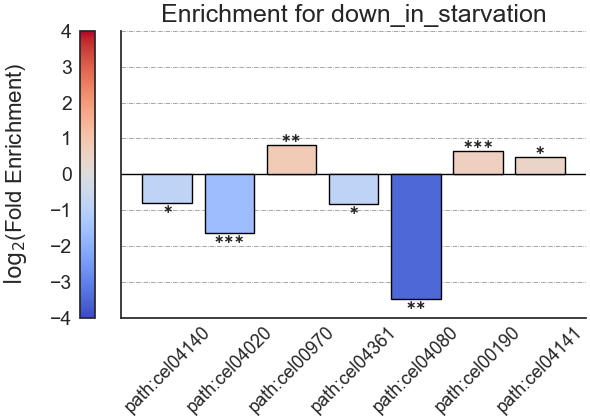
Example plot of kegg_enrichment(plot_horizontal = False)
- map_orthologs_ensembl(map_to_organism: str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'], map_from_organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the Ensembl database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_orthoinspector(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the OrthoInspector database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_panther(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_least_diverged: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PantherDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_least_diverged (bool (default=True)) – if True (default), RNAlysis will only fetch ortholog mappings that were flagged as a ‘least diverged ortholog’ on the PantherDB database. You can read more about this flag on the PantherDB website: https://www.pantherdb.org/genes/
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_phylomedb(map_to_organism: str | int | Literal, map_from_organism: Literal['auto'] | str | int | Literal = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', consistency_score_threshold: Fraction = 0.5, filter_consistency_score: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PhylomeDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- param map_to_organism:
organism name or NCBI taxon ID of the target species for ortholog mapping.
- type map_to_organism:
str or int
- param map_from_organism:
organism name or NCBI taxon ID of the input genes’ source species.
- type map_from_organism:
str or int
- param gene_id_type:
the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- type gene_id_type:
str or ‘auto’ (default=’auto’)
- ` :param consistency_score_threshold: the minimum consistency score required for an ortholog mapping to be considered valid. Consistency scores are calculated by PhylomeDB and represent the confidence of the ortholog mapping. setting consistency_score_threshold to 0 will keep all mappings. You can read more about PhylomeDB consistency score on the PhylomeDB website: orthology.phylomedb.org/help
- type consistency_score_threshold:
float between 0 and 1 (default=0.5)
- param filter_consistency_score:
if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest consistency score.
- type filter_consistency_score:
bool (default=True)
- param non_unique_mode:
How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
- type non_unique_mode:
‘first’, ‘last’, ‘random’, or ‘none’ (default=’first’)
- param remove_unmapped_genes:
if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
- type remove_unmapped_genes:
bool (default=False)
- type inplace:
bool (default=True)
- param inplace:
If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- return:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- non_categorical_enrichment(background_genes: Set[str] | Filter | FeatureSet, attributes: List[str] | str | List[int] | int | Literal['all'], alpha: Fraction = 0.05, parametric_test: bool = False, attr_ref_path: str | Path | Literal['predefined'] = 'predefined', plot_log_scale: bool = True, plot_style: Literal['interleaved', 'overlap'] = 'overlap', n_bins: PositiveInt = 50, save_csv: bool = False, fname=None, return_fig: bool = False, gui_mode: bool = False) DataFrame | Tuple[DataFrame, List[Figure]]
Calculates enrichment and depletion of the FeatureSet for user-defined non-categorical attributes against a background set using either a one-sample T-test or Sign test. The attributes are drawn from an Attribute Reference Table. The background set is determined by either the input variable ‘background_genes’, or by the input variable ‘biotype’ and a Biotype Reference Table. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method).
- Parameters:
attributes (str, int, iterable (list, tuple, set, etc) of str/int, or 'all'.) – An iterable of attribute names or attribute numbers (according to their order in the Attribute Reference Table). If ‘all’, all of the attributes in the Attribute Reference Table will be used. If None, a manual input prompt will be raised.
alpha (float between 0 and 1 (default=0.05)) – Indicates the FDR threshold for significance.
parametric_test (bool (default=False)) – if True, performs a parametric statistical test (one-sample t-test). If False (default), performs a non-parametric statistical test (sign test).
background_genes (set of feature indices, filtering.Filter object, or enrichment.FeatureSet object) – a set of specific feature indices to be used as background genes.
attr_ref_path (str or pathlib.Path (default='predefined')) – the path of the Attribute Reference Table from which user-defined attributes will be drawn.
plot_log_scale (bool (default=True)) – if True (default), the Y-axis of the enrichment plot will be logarithmic. Otherwise, the Y-axis of the enrichment plot will be linear.
plot_style ('overlap' or 'interleaved' (default='overlap')) – ‘interleaved’ will plot an interleaved histogram. ‘overlap’ will plot a semi-transparent histogram where the obsreved and expected are overlapping.
n_bins (int larger than 0 (default=50)) – the number of bins to display in the enrichment plot histograms
save_csv (bool (default=False)) – If True, will save the results to a .csv file, under the name specified in ‘fname’.
fname (str or pathlib.Path (default=None)) – The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt.
return_fig (bool (default=False)) – if True, returns a matplotlib Figure object in addition to the results DataFrame.
- Return type:
pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure]
- Returns:
a polars DataFrame with the indicated attribute names in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
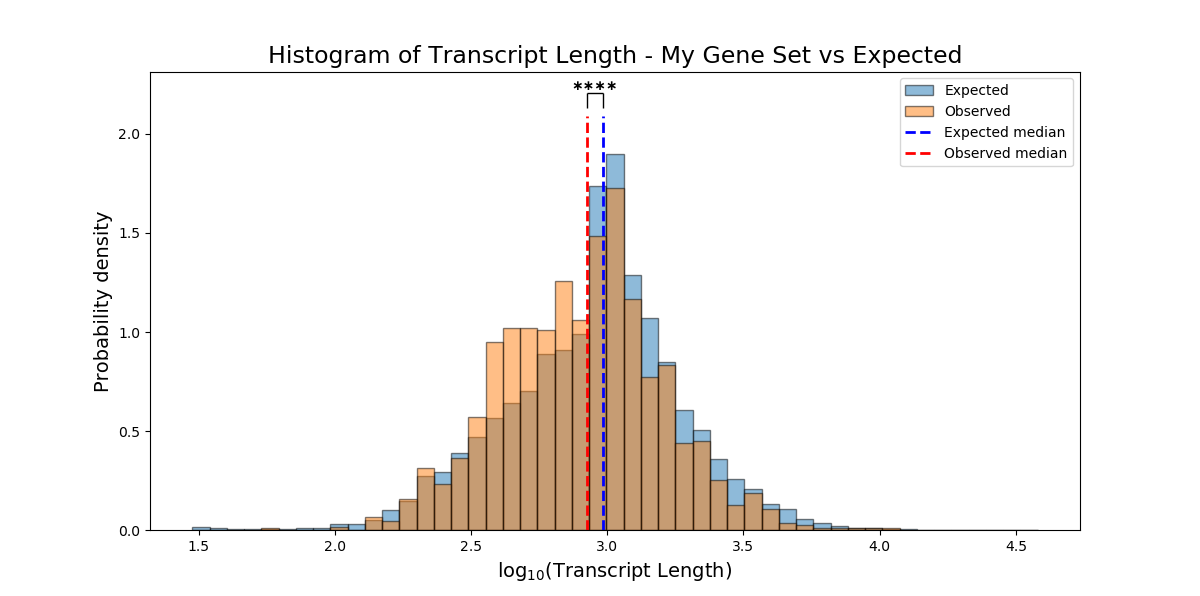
Example plot of non_categorical_enrichment(plot_style`=’overlap’)
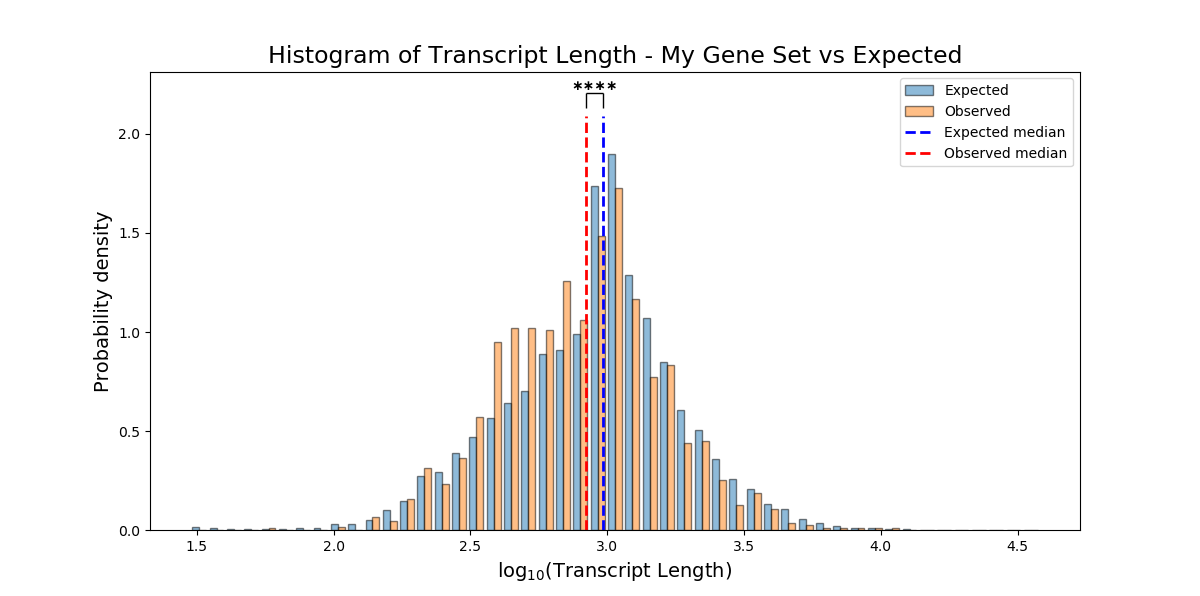
Example plot of non_categorical_enrichment(plot_style=’interleaved’)
- pop()
Remove and return an arbitrary set element. Raises KeyError if the set is empty.
- remove()
Remove an element from a set; it must be a member.
If the element is not a member, raise a KeyError.
- save_txt(fname: str | Path)
Save the list of features in the FeatureSet object under the specified filename and path.
- Parameters:
fname (str or pathlib.Path) – full filename/path for the output file. Can include the ‘.txt’ suffix but doesn’t have to.
- set_name
name of the FeatureSet
- symmetric_difference(other: set | FeatureSet) FeatureSet
Calculates the set symmetric difference of the indices from two FeatureSet objects (the indices that appear in EXACTLY ONE of the FeatureSet objects, and not both/neither). A-symmetric difference-B is equivalent to (A-difference-B)-union-(B-difference-A).
- Parameters:
other (FeatureSet, RankedSet or set) – A second object against which the current object will be compared.
- Returns:
a new FeatureSet with elements in either this FeatureSet or the other object, but not both.
- Return type:
- Examples:
>>> from rnalysis import enrichment >>> en = enrichment.FeatureSet({'WBGene00000001','WBGene00000002','WBGene00000006'}, 'set name') >>> en2 = enrichment.FeatureSet({'WBGene00000004','WBGene00000001'}) >>> en.symmetric_difference(en2) >>> print(en) FeatureSet: set name {'WBGene00000002', 'WBGene00000006', 'WBGene00000004'}
- symmetric_difference_update()
Update a set with the symmetric difference of itself and another.
- translate_gene_ids(translate_to: str | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'], translate_from: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', remove_unmapped_genes: bool = False, inplace: bool = True) FeatureSet
Translates gene names/IDs from one type to another. Mapping is done using the UniProtKB Gene ID Mapping service. You can choose to optionally drop from the table all rows that failed to be translated.
- Parameters:
translate_to (str) – the gene ID type to translate gene names/IDs to. For example: UniProtKB, Ensembl, Wormbase.
translate_from (str or 'auto' (default='auto')) – the gene ID type to translate gene names/IDs from. For example: UniProtKB, Ensembl, Wormbase. If translate_from=’auto’, RNAlysis will attempt to automatically determine the gene ID type of the features in the table.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be translated will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
- Returns:
returns a new and translated FeatureSet.
- union(*others: set | FeatureSet) FeatureSet
Calculates the set union of the indices from multiple FeatureSet objects (the indices that exist in at least one of the FeatureSet objects).
- Parameters:
others (FeatureSet or set) – The objects against which the current object will be compared.
- Returns:
a new FeatureSet with elements from this FeatureSet and all other objects.
- Return type:
- Examples:
>>> from rnalysis import enrichment >>> en = enrichment.FeatureSet({'WBGene00000004','WBGene00000005','WBGene00000006'}, 'set name') >>> en2 = enrichment.FeatureSet({'WBGene00000004','WBGene00000001'}) >>> s = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> en.union(s, en2) >>> print(en) FeatureSet: set name {'WBGene00000003', 'WBGene00000004', 'WBGene00000001', 'WBGene00000002', 'WBGene00000006', 'WBGene00000005'}
- update()
Update a set with the union of itself and others.
- user_defined_enrichment(background_genes: Set[str] | Filter | FeatureSet, attributes: List[str] | str | List[int] | int | Literal['all'], statistical_test: Literal['fisher', 'hypergeometric', 'randomization'] = 'fisher', alpha: Fraction = 0.05, attr_ref_path: str | Path | Literal['predefined'] = 'predefined', exclude_unannotated_genes: bool = True, return_nonsignificant: bool = True, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', randomization_reps: PositiveInt = 10000, random_seed: int | None = None, parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) DataFrame | Tuple[DataFrame, Figure]
Calculates enrichment and depletion of the FeatureSet for user-defined attributes against a background set. The attributes are drawn from an Attribute Reference Table. The background set is determined by either the input variable ‘background_genes’, or by the input variable ‘biotype’ and a Biotype Reference Table. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment score = 0) appears with the smallest value in the scale.
- Parameters:
attributes (str, int, iterable (list, tuple, set, etc) of str/int, or 'all'.) – An iterable of attribute names or attribute numbers (according to their order in the Attribute Reference Table). If ‘all’, all of the attributes in the Attribute Reference Table will be used. If None, a manual input prompt will be raised.
statistical_test ('fisher', 'hypergeometric' or 'randomization' (default='fisher')) – determines the statistical test to be used for enrichment analysis. Note that some propagation methods support only some of the available statistical tests.
alpha (float between 0 and 1 (default=0.05)) – Indicates the FDR threshold for significance.
attr_ref_path (str or pathlib.Path (default='predefined')) – the path of the Attribute Reference Table from which user-defined attributes will be drawn.
biotype_ref_path (str or pathlib.Path (default='predefined')) – the path of the Biotype Reference Table. Will be used to generate background set if ‘biotype’ is specified.
biotype (str specifying a specific biotype, list/set of strings each specifying a biotype, or 'all' (default='protein_coding')) – determines the background genes by their biotype. Requires specifying a Biotype Reference Table. ‘all’ will include all genomic features in the reference table, ‘protein_coding’ will include only protein-coding genes from the reference table, etc. Cannot be specified together with ‘background_genes’.
background_genes (set of feature indices, filtering.Filter object, or enrichment.FeatureSet object) – a set of specific feature indices to be used as background genes.
exclude_unannotated_genes (bool (deafult=True)) – if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results.
return_nonsignificant (bool (default=True)) – if True (default), the results DataFrame will include all tested attributes - both significant and non-significant ones. If False, only significant attributes will be returned.
save_csv (bool (default=False)) – If True, will save the results to a .csv file, under the name specified in ‘fname’.
fname (str or pathlib.Path (default=None)) – The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt.
return_fig (bool (default=False)) – if True, returns a matplotlib Figure object in addition to the results DataFrame.
plot_horizontal (bool (default=True)) – if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot.
show_expected (bool (default=False)) – if True, the observed/expected values will be shown on the plot.
plot_style ('bar' or 'lollipop' (default='bar')) – style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set.
randomization_reps (int larger than 0 (default=10000)) – if using a randomization test, determine how many randomization repititions to run. Otherwise, this parameter will not affect the analysis.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Return type:
pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure]
- Returns:
a polars DataFrame with the indicated attribute names in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
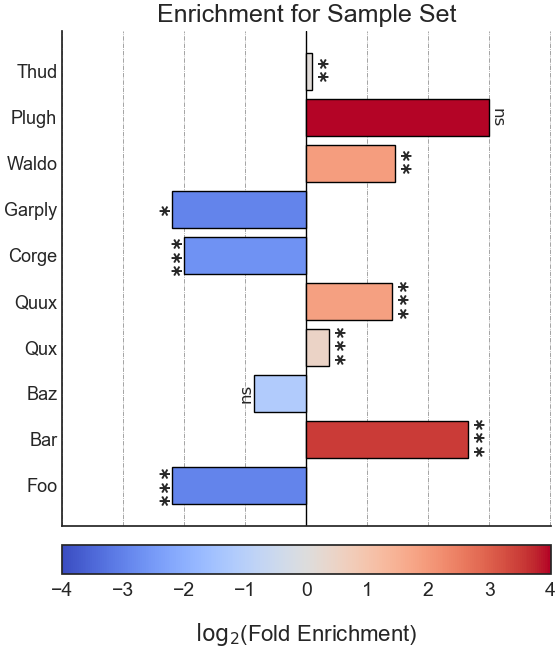
Example plot of user_defined_enrichment()
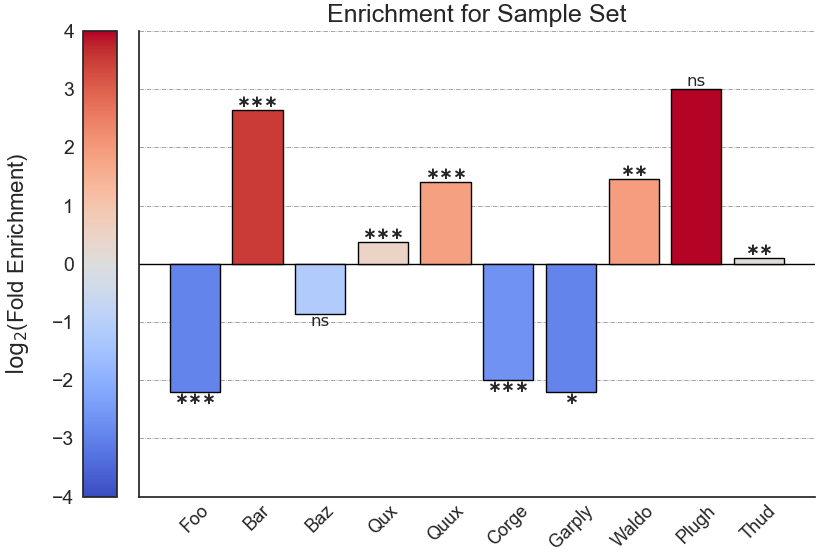
Example plot of user_defined_enrichment(plot_horizontal = False)
- class rnalysis.enrichment.RankedSet(ranked_genes: Filter | List[str] | Tuple[str] | ndarray, set_name: str = '')
Bases:
FeatureSetReceives a ranked gene set, sorted by any biologically-meaningful value (expression level, fold-change, etc) and preforms various enrichment analyses on them. ALl functions that can be applied to FeatureSet objects can also be applied to RankedSet objects.
- _inplace(func, func_kwargs, inplace: bool, **update_kwargs)
Executes the user’s choice whether to filter in-place or create a new instance of the FeatureSet object.
- _set_ops(others: set | FeatureSet, op: LambdaType)
Performs a given set operation on self and on another object (FeatureSet or set). :type others: FeatureSet or set :param others: Other object to perform set operation with. :type: op: Callable (set.union, set.intersection, set.difference or set.symmetric difference) :param op: The set operation to be performed. :return: A set resulting from the set operation.
- add()
Add an element to a set.
This has no effect if the element is already present.
- biotypes_from_gtf(gtf_path: str | Path, attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene') DataFrame
Returns a DataFrame describing the biotypes in the table and their count. The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
:param long_format:if True, returns a short-form DataFrame, which states the biotypes in the Filter object and their count. Otherwise, returns a long-form DataFrame, which also provides descriptive statistics of each column per biotype. :rtype: pl.DataFrame :returns: a polars DataFrame showing the number of values belonging to each biotype, as well as additional descriptive statistics of format==’long’.
- biotypes_from_ref_table(ref: str | Path | Literal['predefined'] = 'predefined')
Returns a DataFrame describing the biotypes in the gene set and their count.
- Parameters:
ref (str or pathlib.Path (default='predefined')) – Path of the reference file used to determine biotype. Default is the path predefined in the settings file.
- Examples:
>>> from rnalysis import enrichment, filtering >>> d = filtering.Filter("tests/test_files/test_deseq.csv") >>> en = enrichment.FeatureSet(d) >>> en.biotypes_from_ref_table(ref='tests/test_files/biotype_ref_table_for_tests.csv') Biotype Reference Table used: tests/test_files/biotype_ref_table_for_tests.csv shape: (3, 2) ┌────────────────┬───────┐ │ biotype ┆ count │ │ --- ┆ --- │ │ str ┆ u32 │ ╞════════════════╪═══════╡ │ protein_coding ┆ 26 │ │ pseudogene ┆ 1 │ │ unknown ┆ 1 │ └────────────────┴───────┘
- change_set_name(new_name: str)
Change the ‘set_name’ of a FeatureSet to a new name.
- Parameters:
new_name (str) – the new set name
- clear()
Remove all elements from this set.
- copy()
Return a shallow copy of a set.
- difference(*others: set | FeatureSet) FeatureSet
Calculates the set difference of the indices from multiple FeatureSet objects (the indices that appear in the first FeatureSet object but NOT in the other objects).
- Parameters:
others (FeatureSet, RankedSet or set) – The objects against which the current object will be compared.
- Returns:
a new FeatureSet with elements in this FeatureSet that are not in the other objects.
- Return type:
- Examples:
>>> from rnalysis import enrichment >>> en = enrichment.FeatureSet({'WBGene00000001','WBGene00000002','WBGene00000006'}, 'set name') >>> s = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> en2 = enrichment.FeatureSet({'WBGene00000004','WBGene00000001'}) >>> en.difference(s, en2) >>> print(en) FeatureSet: set name {'WBGene00000006'}
- difference_update()
Remove all elements of another set from this set.
- discard()
Remove an element from a set if it is a member.
Unlike set.remove(), the discard() method does not raise an exception when an element is missing from the set.
- filter_biotype_from_gtf(gtf_path: str | Path, biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', attribute_name: Literal['biotype', 'gene_biotype', 'transcript_biotype', 'gene_type', 'transcript_type'] | str = 'gene_biotype', feature_type: Literal['gene', 'transcript'] = 'gene', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a GTF (Gene transfer format) file supplied by the user.
- Parameters:
gtf_path (str or Path) – Path to your GTF (Gene transfer format) file. The file should match the type of gene names/IDs you use in your table, and should contain an attribute describing biotype.
biotype (str or list of strings) – the biotypes which will not be filtered out.
attribute_name (str (default='gene_biotype')) – name of the attribute in your GTF file that describes feature biotype.
feature_type ('gene' or 'transcript' (default='gene')) – determined whether the features/rows in your data table describe individual genes or transcripts.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- filter_biotype_from_ref_table(biotype: Literal['protein_coding', 'pseudogene', 'lincRNA', 'miRNA', 'ncRNA', 'piRNA', 'rRNA', 'snoRNA', 'snRNA', 'tRNA'] | str | List[str] = 'protein_coding', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters out all features that do not match the indicated biotype/biotypes (for example: ‘protein_coding’, ‘ncRNA’, etc). The data about feature biotypes is drawn from a Biotype Reference Table supplied by the user.
- Parameters:
biotype (string or list of strings) – the biotypes which will not be filtered out.
ref – Name of the biotype reference file used to determine biotypes. Default is the path defined by the user in the settings.yaml file.
opposite (bool) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- filter_by_attribute(attributes: str | List[str], mode: Literal['union', 'intersection'] = 'union', ref: str | Path | Literal['predefined'] = 'predefined', opposite: bool = False, inplace: bool = True)
Filters features according to user-defined attributes from an Attribute Reference Table. When multiple attributes are given, filtering can be done in ‘union’ mode (where features that belong to at least one attribute are not filtered out), or in ‘intersection’ mode (where only features that belong to ALL attributes are not filtered out). To learn more about user-defined attributes and Attribute Reference Tables, read the user guide.
- Parameters:
attributes (string or list of strings, which are column titles in the user-defined Attribute Reference Table.) – attributes to filter by.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
ref (str or pathlib.Path (default='predefined')) – filename/path of the attribute reference table to be used as reference.
opposite (bool (default=False)) – If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
If ‘inplace’ is False, returns a new and filtered instance of the Filter object.
- filter_by_go_annotations(go_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', propagate_annotations: bool = True, evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', opposite: bool = False, inplace: bool = True)
Filters genes according to GO annotations, keeping only genes that are annotated with a specific GO term. When multiple GO terms are given, filtering can be done in ‘union’ mode (where genes that belong to at least one GO term are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL GO terms are not filtered out).
- Parameters:
go_ids (str or list of str)
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :param propagate_annotations: determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/ :type propagate_annotations: ‘classic’, ‘elim’, ‘weight’, ‘all.m’, or ‘no’ (default=’elim’) :param evidence_types: only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or ‘any’ (default=’any’) :param excluded_evidence_types: annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/ :type excluded_evidence_types: str, Iterable of str, ‘experimental’, ‘phylogenetic’ ,’computational’, ‘author’, ‘curator’, ‘electronic’, or None (default=None) :param databases: only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current FeatureSet object. If False, the function will return a new FeatureSet instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the FeatureSet object.
- filter_by_kegg_annotations(kegg_ids: str | List[str], mode: Literal['union', 'intersection'] = 'union', organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', opposite: bool = False, inplace: bool = True)
Filters genes according to KEGG pathways, keeping only genes that belong to specific KEGG pathway. When multiple KEGG IDs are given, filtering can be done in ‘union’ mode (where genes that belong to at least one pathway are not filtered out), or in ‘intersection’ mode (where only genes that belong to ALL pathways are not filtered out).
- Parameters:
kegg_ids (str or list of str) – the KEGG pathway IDs according to which the table will be filtered. An example for a legal KEGG pathway ID would be ‘path:cel04020’ for the C. elegans calcium signaling pathway.
mode ('union' or 'intersection'.) – If ‘union’, filters out every genomic feature that does not belong to one or more of the indicated attributes. If ‘intersection’, filters out every genomic feature that does not belong to ALL of the indicated attributes.
param organism: organism name or NCBI taxon ID for which the function will fetch GO annotations. :type organism: str or int :param gene_id_type: the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping :type gene_id_type: str or ‘auto’ (default=’auto’) :type opposite: bool (default=False) :param opposite: If True, the output of the filtering will be the OPPOSITE of the specified (instead of filtering out X, the function will filter out anything BUT X). If False (default), the function will filter as expected. :type inplace: bool (default=True) :param inplace: If True (default), filtering will be applied to the current FeatureSet object. If False, the function will return a new Filter instance and the current instance will not be affected. :return: If ‘inplace’ is False, returns a new, filtered instance of the FeatureSet object.
- find_paralogs_ensembl(organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True)
Find paralogs within the same species using the Ensembl database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
- Returns:
DataFrame describing all discovered paralog mappings.
- find_paralogs_panther(organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto')
Find paralogs within the same species using the PantherDB database.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- Returns:
DataFrame describing all discovered paralog mappings.
- gene_set
- go_enrichment(background_genes: Set[str] | Filter | FeatureSet, organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', alpha: Fraction = 0.05, statistical_test: Literal['fisher', 'hypergeometric', 'randomization'] = 'fisher', propagate_annotations: Literal['classic', 'elim', 'weight', 'all.m', 'no'] = 'elim', aspects: Literal['any', 'biological_process', 'cellular_component', 'molecular_function'] | Iterable[Literal['biological_process', 'cellular_component', 'molecular_function']] = 'any', evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] | Literal['any'] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', exclude_unannotated_genes: bool = True, return_nonsignificant: bool = False, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', plot_ontology_graph: bool = True, ontology_graph_format: Literal['pdf', 'png', 'svg', 'none'] = 'none', randomization_reps: PositiveInt = 10000, random_seed: int | None = None, parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) DataFrame | Tuple[DataFrame, Figure]
Calculates enrichment and depletion of the FeatureSet for Gene Ontology (GO) terms against a background set. The GO terms and annotations are drawn via the GO Solr search engine GOlr, using the search terms defined by the user. The background set is determined by either the input variable ‘background_genes’, or by the input variable ‘biotype’ and a Biotype Reference Table. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment score = 0) appears with the smallest value in the scale.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID for which the function will fetch GO annotations.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the GOLR server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
alpha (float between 0 and 1 (default=0.05)) – Indicates the FDR threshold for significance.
statistical_test ('fisher', 'hypergeometric' or 'randomization' (default='fisher')) – determines the statistical test to be used for enrichment analysis. Note that some propagation methods support only some of the available statistical tests.
biotype (str specifying a specific biotype, list/set of strings each specifying a biotype, or 'all'. Default 'protein_coding'.) – determines the background genes by their biotype. Requires specifying a Biotype Reference Table. ‘all’ will include all genomic features in the reference table, ‘protein_coding’ will include only protein-coding genes from the reference table, etc. Cannot be specified together with ‘background_genes’.
background_genes (set of feature indices, filtering.Filter object, or enrichment.FeatureSet object) – a set of specific feature indices to be used as background genes.
biotype_ref_path (str or pathlib.Path (default='predefined')) – the path of the Biotype Reference Table. Will be used to generate background set if ‘biotype’ is specified.
propagate_annotations ('classic', 'elim', 'weight', 'all.m', or 'no' (default='elim')) – determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/
aspects (str, Iterable of str, 'biological_process', 'molecular_function', 'cellular_component', or 'any' (default='any')) – only annotations from the specified GO aspects will be included in the analysis. Legal aspects are ‘biological_process’ (P), ‘molecular_function’ (F), and ‘cellular_component’ (C).
evidence_types (str, Iterable of str, 'experimental', 'phylogenetic' ,'computational', 'author', 'curator', 'electronic', or 'any' (default='any')) – only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/
excluded_evidence_types (str, Iterable of str, 'experimental', 'phylogenetic' ,'computational', 'author', 'curator', 'electronic', or None (default=None)) – annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/
databases – only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website:
http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :param exclude_unannotated_genes: if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results. :type exclude_unannotated_genes: bool (deafult=True) :param return_nonsignificant: if True, the results DataFrame will include all tested GO terms - both significant and non-significant terms. If False (default), only significant GO terms will be returned. :type return_nonsignificant: bool (default=False) :type save_csv: bool, default False :param save_csv: If True, will save the results to a .csv file, under the name specified in ‘fname’. :type fname: str or pathlib.Path :param fname: The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt. :type return_fig: bool (default=False) :param return_fig: if True, returns a matplotlib Figure object in addition to the results DataFrame. :type plot_ontology_graph: bool (default=True) :param plot_ontology_graph: if True, will generate an ontology graph depicting the significant GO terms and their parent nodes. :type ontology_graph_format: ‘pdf’, ‘png’, ‘svg’, or ‘none’ (default=’none’) :param ontology_graph_format: if ontology_graph_format is not ‘none’, the ontology graph will additonally be generated in the specified file format. :type plot_horizontal: bool (default=True) :param plot_horizontal: if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot. :param show_expected: if True, the observed/expected values will be shown on the plot. :type show_expected: bool (default=False) :param plot_style: style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set. :type plot_style: ‘bar’ or ‘lollipop’ (default=’bar’) :type random_seed: non-negative integer (default=None) :type random_seed: if using a randomization test, determine the random seed used to initialize the pseudorandom generator for the randomization test. By default it is picked at random, but you can set it to a particular integer to get consistents results over multiple runs. If not using a randomization test, this parameter will not affect the analysis. :param randomization_reps: if using a randomization test, determine how many randomization repititions to run. Otherwise, this parameter will not affect the analysis. :type randomization_reps: int larger than 0 (default=10000) :type parallel_backend: Literal[PARALLEL_BACKENDS] (default=’loky’) :param parallel_backend: Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise. :rtype: pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure] :return: a polars DataFrame with GO terms in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
Example plot of go_enrichment(plot_ontology_graph=True)
Example plot of go_enrichment()
Example plot of go_enrichment(plot_horizontal = False)
- intersection(*others: set | FeatureSet) FeatureSet
Calculates the set intersection of the indices from multiple FeatureSet objects (the indices that exist in ALL of the FeatureSet objects).
- Parameters:
others (FeatureSet, RankedSet or set) – The objects against which the current object will be compared.
- Returns:
a new FeatureSet with elements common to this FeatureSet and all other objects.
- Return type:
- Examples:
>>> from rnalysis import enrichment >>> en = enrichment.FeatureSet({'WBGene00000001','WBGene00000002','WBGene00000006'}, 'set name') >>> s = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> en2 = enrichment.FeatureSet({'WBGene00000004','WBGene00000001'}) >>> en.intersection(s, en2) >>> print(en) FeatureSet: set name {'WBGene00000001'}
- intersection_update()
Update a set with the intersection of itself and another.
- isdisjoint()
Return True if two sets have a null intersection.
- issubset(other, /)
Test whether every element in the set is in other.
- issuperset(other, /)
Test whether every element in other is in the set.
- kegg_enrichment(background_genes: Set[str] | Filter | FeatureSet, organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', alpha: Fraction = 0.05, statistical_test: Literal['fisher', 'hypergeometric', 'randomization'] = 'fisher', exclude_unannotated_genes: bool = True, return_nonsignificant: bool = False, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', plot_pathway_graphs: bool = True, pathway_graphs_format: Literal['pdf', 'png', 'svg', 'none'] = 'none', randomization_reps: PositiveInt = 10000, random_seed: int | None = None, parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) DataFrame | Tuple[DataFrame, Figure]
Calculates enrichment and depletion of the FeatureSet for Kyoto Encyclopedia of Genes and Genomes (KEGG) curated pathways against a background set. The background set is determined by either the input variable ‘background_genes’, or by the input variable ‘biotype’ and a Biotype Reference Table. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment score = 0) appears with the smallest value in the scale.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID for which the function will fetch GO annotations.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
alpha (float between 0 and 1 (default=0.05)) – Indicates the FDR threshold for significance.
statistical_test ('fisher', 'hypergeometric' or 'randomization' (default='fisher')) – determines the statistical test to be used for enrichment analysis. Note that some propagation methods support only some of the available statistical tests.
background_genes (set of feature indices, filtering.Filter object, or enrichment.FeatureSet object) – a set of specific feature indices to be used as background genes.
exclude_unannotated_genes (bool (deafult=True)) – if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results.
return_nonsignificant (bool (default=False)) – if True, the results DataFrame will include all tested pathways - both significant and non-significant ones. If False (default), only significant pathways will be returned.
save_csv (bool, default False) – If True, will save the results to a .csv file, under the name specified in ‘fname’.
fname (str or pathlib.Path) – The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt.
return_fig (bool (default=False)) – if True, returns a matplotlib Figure object in addition to the results DataFrame.
plot_horizontal (bool (default=True)) – if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot.
show_expected (bool (default=False)) – if True, the observed/expected values will be shown on the plot.
plot_style ('bar' or 'lollipop' (default='bar')) – style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set.
plot_pathway_graphs (bool (default=True)) – if True, will generate pathway graphs depicting the significant KEGG pathways.
pathway_graphs_format ('pdf', 'png', 'svg', or None (default=None)) – if pathway_graphs_format is not ‘none’, the pathway graphs will additonally be generated in the specified file format.
randomization_reps (int larger than 0 (default=10000)) – if using a randomization test, determine how many randomization repititions to run. Otherwise, this parameter will not affect the analysis.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Return type:
pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure]
- Returns:
a polars DataFrame with the indicated pathway names in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
Example plot of kegg_enrichment()
Example plot of kegg_enrichment(plot_horizontal = False)
- map_orthologs_ensembl(map_to_organism: str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'], map_from_organism: Literal['auto'] | str | int | Literal['Acanthochromis polyacanthus', 'Accipiter nisus', 'Ailuropoda melanoleuca', 'Amazona collaria', 'Amphilophus citrinellus', 'Amphiprion ocellaris', 'Amphiprion percula', 'Anabas testudineus', 'Anas platyrhynchos', 'Anas platyrhynchos platyrhynchos', 'Anas zonorhyncha', 'Anolis carolinensis', 'Anser brachyrhynchus', 'Anser cygnoides', 'Aotus nancymaae', 'Apteryx haastii', 'Apteryx owenii', 'Apteryx rowi', 'Aquila chrysaetos chrysaetos', 'Astatotilapia calliptera', 'Astyanax mexicanus', 'Astyanax mexicanus pachon', 'Athene cunicularia', 'Balaenoptera musculus', 'Betta splendens', 'Bison bison bison', 'Bos grunniens', 'Bos indicus hybrid', 'Bos mutus', 'Bos taurus', 'Bos taurus gca963921495v1', 'Bos taurus hybrid', 'Bos taurus tuli', 'Bos taurus wagyu', 'Bubo bubo', 'Buteo japonicus', 'Cairina moschata domestica', 'Calidris pugnax', 'Calidris pygmaea', 'Callithrix jacchus', 'Callorhinchus milii', 'Camarhynchus parvulus', 'Camelus dromedarius', 'Canis lupus dingo', 'Canis lupus familiaris', 'Canis lupus familiarisbasenji', 'Canis lupus familiarisboxer', 'Canis lupus familiarisgreatdane', 'Canis lupus familiarisgsd', 'Capra hircus', 'Capra hircus blackbengal', 'Capra hircus gca015443085v1', 'Capra hircus gca026652205v1', 'Carassius auratus', 'Carlito syrichta', 'Castor canadensis', 'Catagonus wagneri', 'Catharus ustulatus', 'Cavia aperea', 'Cavia porcellus', 'Cebus imitator', 'Cercocebus atys', 'Cervus hanglu yarkandensis', 'Chelonoidis abingdonii', 'Chelydra serpentina', 'Chinchilla lanigera', 'Chlorocebus sabaeus', 'Choloepus hoffmanni', 'Chrysemys picta bellii', 'Chrysolophus pictus', 'Ciona intestinalis', 'Ciona savignyi', 'Clupea harengus', 'Colobus angolensis palliatus', 'Corvus moneduloides', 'Cottoperca gobio', 'Coturnix japonica', 'Cricetulus griseus chok1gshd', 'Cricetulus griseus crigri', 'Cricetulus griseus picr', 'Crocodylus porosus', 'Cyanistes caeruleus', 'Cyclopterus lumpus', 'Cynoglossus semilaevis', 'Cyprinodon variegatus', 'Cyprinus carpio carpio', 'Cyprinus carpio germanmirror', 'Cyprinus carpio hebaored', 'Cyprinus carpio huanghe', 'Danio rerio', 'Dasypus novemcinctus', 'Delphinapterus leucas', 'Denticeps clupeoides', 'Dicentrarchus labrax', 'Dipodomys ordii', 'Dromaius novaehollandiae', 'Echeneis naucrates', 'Echinops telfairi', 'Electrophorus electricus', 'Eptatretus burgeri', 'Equus asinus', 'Equus asinus gca041296235v2', 'Equus caballus', 'Erinaceus europaeus', 'Erpetoichthys calabaricus', 'Erythrura gouldiae', 'Esox lucius', 'Falco tinnunculus', 'Felis catus', 'Felis catus abyssinian', 'Ficedula albicollis', 'Fukomys damarensis', 'Fundulus heteroclitus', 'Gadus morhua', 'Gadus morhua gca010882105v1', 'Gallus gallus', 'Gallus gallus gca000002315v5', 'Gallus gallus gca016700215v2', 'Gambusia affinis', 'Gasterosteus aculeatus', 'Gasterosteus aculeatus gca006229185v1', 'Gasterosteus aculeatus gca006232265v1', 'Gasterosteus aculeatus gca006232285v1', 'Geospiza fortis', 'Gopherus agassizii', 'Gopherus evgoodei', 'Gorilla gorilla', 'Gouania willdenowi', 'Haplochromis burtoni', 'Heterocephalus glaber female', 'Heterocephalus glaber male', 'Hippocampus comes', 'Homo sapiens', 'Hucho hucho', 'Ictalurus punctatus', 'Ictidomys tridecemlineatus', 'Jaculus jaculus', 'Junco hyemalis', 'Kryptolebias marmoratus', 'Labrus bergylta', 'Larimichthys crocea', 'Lates calcarifer', 'Laticauda laticaudata', 'Latimeria chalumnae', 'Lepidothrix coronata', 'Lepisosteus oculatus', 'Leptobrachium leishanense', 'Lonchura striata domestica', 'Loxodonta africana', 'Lynx canadensis', 'Macaca fascicularis', 'Macaca mulatta', 'Macaca nemestrina', 'Malurus cyaneus samueli', 'Manacus vitellinus', 'Mandrillus leucophaeus', 'Marmota marmota marmota', 'Mastacembelus armatus', 'Maylandia zebra', 'Meleagris gallopavo', 'Melopsittacus undulatus', 'Meriones unguiculatus', 'Mesocricetus auratus', 'Microcebus murinus', 'Microtus ochrogaster', 'Mola mola', 'Monodelphis domestica', 'Monodon monoceros', 'Monopterus albus', 'Moschus moschiferus', 'Mus caroli', 'Mus musculus', 'Mus musculus 129s1svimj', 'Mus musculus aj', 'Mus musculus akrj', 'Mus musculus balbcj', 'Mus musculus c3hhej', 'Mus musculus c57bl6nj', 'Mus musculus casteij', 'Mus musculus cbaj', 'Mus musculus dba2j', 'Mus musculus fvbnj', 'Mus musculus lpj', 'Mus musculus molossinusjf1msj', 'Mus musculus nodshiltj', 'Mus musculus nzohlltj', 'Mus musculus pwkphj', 'Mus musculus wsbeij', 'Mus pahari', 'Mus spicilegus', 'Mus spretus', 'Mustela putorius furo', 'Myotis lucifugus', 'Myripristis murdjan', 'Naja naja', 'Nannospalax galili', 'Neogobius melanostomus', 'Neolamprologus brichardi', 'Neovison vison', 'Nomascus leucogenys', 'Notamacropus eugenii', 'Notechis scutatus', 'Nothobranchius furzeri', 'Nothoprocta perdicaria', 'Numida meleagris', 'Ochotona princeps', 'Octodon degus', 'Oncorhynchus kisutch', 'Oncorhynchus mykiss', 'Oncorhynchus tshawytscha', 'Oreochromis aureus', 'Oreochromis niloticus', 'Ornithorhynchus anatinus', 'Oryctolagus cuniculus', 'Oryzias javanicus', 'Oryzias latipes', 'Oryzias latipes hni', 'Oryzias latipes hsok', 'Oryzias melastigma', 'Oryzias sinensis', 'Otolemur garnettii', 'Otus sunia', 'Ovis aries', 'Ovis aries gca011170295v1', 'Ovis aries gca018804185v1', 'Ovis aries gca022416685v1', 'Ovis aries gca022416695v1', 'Ovis aries gca022416755v1', 'Ovis aries gca022416775v1', 'Ovis aries gca022416915v1', 'Ovis aries gca022432825v1', 'Ovis aries gca022432835v1', 'Ovis aries gca024222175v1', 'Ovis aries texel', 'Pan paniscus', 'Pan troglodytes', 'Panthera leo', 'Panthera pardus', 'Panthera tigris altaica', 'Papio anubis', 'Parambassis ranga', 'Paramormyrops kingsleyae', 'Parus major', 'Pavo cristatus', 'Pelodiscus sinensis', 'Pelusios castaneus', 'Periophthalmus magnuspinnatus', 'Peromyscus maniculatus bairdii', 'Petromyzon marinus', 'Phascolarctos cinereus', 'Phasianus colchicus', 'Phocoena sinus', 'Physeter catodon', 'Piliocolobus tephrosceles', 'Podarcis muralis', 'Poecilia formosa', 'Poecilia latipinna', 'Poecilia mexicana', 'Poecilia reticulata', 'Pogona vitticeps', 'Pongo abelii', 'Procavia capensis', 'Prolemur simus', 'Propithecus coquereli', 'Pseudonaja textilis', 'Pteropus vampyrus', 'Pundamilia nyererei', 'Pygocentrus nattereri', 'Rattus norvegicus', 'Rattus norvegicus shrspbbbutx', 'Rattus norvegicus shrutx', 'Rattus norvegicus wkybbb', 'Rhinolophus ferrumequinum', 'Rhinopithecus bieti', 'Rhinopithecus roxellana', 'Saimiri boliviensis boliviensis', 'Salarias fasciatus', 'Salmo salar', 'Salmo salar gca021399835v1', 'Salmo salar gca923944775v1', 'Salmo salar gca931346935v2', 'Salmo trutta', 'Salvator merianae', 'Sander lucioperca', 'Sarcophilus harrisii', 'Sciurus vulgaris', 'Scleropages formosus', 'Scophthalmus maximus', 'Serinus canaria', 'Seriola dumerili', 'Seriola lalandi dorsalis', 'Sinocyclocheilus anshuiensis', 'Sinocyclocheilus grahami', 'Sinocyclocheilus rhinocerous', 'Sorex araneus', 'Sparus aurata', 'Spermophilus dauricus', 'Sphaeramia orbicularis', 'Sphenodon punctatus', 'Stachyris ruficeps', 'Stegastes partitus', 'Strigops habroptila', 'Strix occidentalis caurina', 'Struthio camelus australis', 'Suricata suricatta', 'Sus scrofa', 'Sus scrofa bamei', 'Sus scrofa berkshire', 'Sus scrofa gca007644095v1', 'Sus scrofa gca015776825v1', 'Sus scrofa gca017957985v1', 'Sus scrofa gca018555405v1', 'Sus scrofa gca019290145v1', 'Sus scrofa gca020567905v1', 'Sus scrofa gca021656055v1', 'Sus scrofa gca024718415v1', 'Sus scrofa gca037447515v1', 'Sus scrofa gca039654815v1', 'Sus scrofa gca040869115v1', 'Sus scrofa gca041937265v1', 'Sus scrofa gca044906105v1', 'Sus scrofa gca044906185v1', 'Sus scrofa gca046128825v1', 'Sus scrofa gca048338725v1', 'Sus scrofa gca963921485v1', 'Sus scrofa hampshire', 'Sus scrofa jinhua', 'Sus scrofa landrace', 'Sus scrofa largewhite', 'Sus scrofa meishan', 'Sus scrofa pietrain', 'Sus scrofa rongchang', 'Sus scrofa tibetan', 'Sus scrofa usmarc', 'Sus scrofa wuzhishan', 'Taeniopygia guttata', 'Takifugu rubripes', 'Terrapene carolina triunguis', 'Tetraodon nigroviridis', 'Theropithecus gelada', 'Tupaia belangeri', 'Tursiops truncatus', 'Urocitellus parryii', 'Ursus americanus', 'Ursus maritimus', 'Ursus thibetanus thibetanus', 'Varanus komodoensis', 'Vicugna pacos', 'Vombatus ursinus', 'Vulpes vulpes', 'Xenopus tropicalis', 'Xiphophorus couchianus', 'Xiphophorus maculatus', 'Zalophus californianus', 'Zonotrichia albicollis', 'Zosterops lateralis melanops'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_percent_identity: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the Ensembl database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_percent_identity (bool (default=True)) – if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest percent_identity score.
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_orthoinspector(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the OrthoInspector database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_panther(map_to_organism: str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'], map_from_organism: Literal['auto'] | str | int | Literal['Amborella trichopoda', 'Anolis carolinensis', 'Anopheles gambiae', 'Aquifex aeolicus', 'Arabidopsis thaliana', 'Bacillus cereus', 'Bacillus subtilis', 'Bacteroides thetaiotaomicron', 'Batrachochytrium dendrobatidis', 'Bos taurus', 'Brachypodium distachyon', 'Bradyrhizobium diazoefficiens', 'Branchiostoma floridae', 'Brassica campestris', 'Brassica napus', 'Caenorhabditis briggsae', 'Caenorhabditis elegans', 'Candida albicans', 'Canis lupus familiaris', 'Capsicum annuum', 'Chlamydia trachomatis', 'Chlamydomonas reinhardtii', 'Chloroflexus aurantiacus', 'Ciona intestinalis', 'Citrus sinensis', 'Clostridium botulinum', 'Coxiella burnetii', 'Cryptococcus neoformans', 'Cucumis sativus', 'Danio rerio', 'Daphnia pulex', 'Deinococcus radiodurans', 'Dictyoglomus turgidum', 'Dictyostelium discoideum', 'Dictyostelium purpureum', 'Drosophila melanogaster', 'Emericella nidulans', 'Entamoeba histolytica', 'Equus caballus', 'Eremothecium gossypii', 'Erythranthe guttata', 'Escherichia coli', 'Eucalyptus grandis', 'Felis catus', 'Fusobacterium nucleatum', 'Gallus gallus', 'Geobacter sulfurreducens', 'Giardia intestinalis', 'Gloeobacter violaceus', 'Glycine max', 'Gorilla gorilla gorilla', 'Gossypium hirsutum', 'Haemophilus influenzae', 'Halobacterium salinarum', 'Helianthus annuus', 'Helicobacter pylori', 'Homo sapiens', 'Hordeum vulgare subsp. vulgare', 'Ixodes scapularis', 'Juglans regia', 'Klebsormidium nitens', 'Korarchaeum cryptofilum', 'Lactuca sativa', 'Leishmania major', 'Leptospira interrogans', 'Listeria monocytogenes', 'Macaca mulatta', 'Manihot esculenta', 'Marchantia polymorpha', 'Medicago truncatula', 'Methanocaldococcus jannaschii', 'Methanosarcina acetivorans', 'Monodelphis domestica', 'Monosiga brevicollis', 'Mus musculus', 'Musa acuminata subsp. malaccensis', 'Mycobacterium tuberculosis', 'Neisseria meningitidis serogroup b', 'Nelumbo nucifera', 'Nematostella vectensis', 'Neosartorya fumigata', 'Neurospora crassa', 'Nicotiana tabacum', 'Nitrosopumilus maritimus', 'Ornithorhynchus anatinus', 'Oryza sativa', 'Oryzias latipes', 'Pan troglodytes', 'Paramecium tetraurelia', 'Phaeosphaeria nodorum', 'Physcomitrella patens', 'Phytophthora ramorum', 'Plasmodium falciparum', 'Populus trichocarpa', 'Pristionchus pacificus', 'Prunus persica', 'Pseudomonas aeruginosa', 'Puccinia graminis', 'Pyrobaculum aerophilum', 'Rattus norvegicus', 'Rhodopirellula baltica', 'Ricinus communis', 'Saccharomyces cerevisiae', 'Salmonella typhimurium', 'Schizosaccharomyces japonicus', 'Schizosaccharomyces pombe', 'Sclerotinia sclerotiorum', 'Selaginella moellendorffii', 'Setaria italica', 'Shewanella oneidensis', 'Solanum lycopersicum', 'Solanum tuberosum', 'Sorghum bicolor', 'Spinacia oleracea', 'Staphylococcus aureus', 'Streptococcus pneumoniae', 'Streptomyces coelicolor', 'Strongylocentrotus purpuratus', 'Sulfolobus solfataricus', 'Sus scrofa', 'Synechocystis', 'Thalassiosira pseudonana', 'Theobroma cacao', 'Thermococcus kodakaraensis', 'Thermodesulfovibrio yellowstonii', 'Thermotoga maritima', 'Tribolium castaneum', 'Trichomonas vaginalis', 'Trichoplax adhaerens', 'Triticum aestivum', 'Trypanosoma brucei', 'Ustilago maydis', 'Vibrio cholerae', 'Vitis vinifera', 'Xanthomonas campestris', 'Xenopus laevis', 'Xenopus tropicalis', 'Yarrowia lipolytica', 'Yersinia pestis', 'Zea mays', 'Zostera marina', 'helobdella robusta', 'lepisosteus oculatus', 'mycoplasma genitalium'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', filter_least_diverged: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PantherDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- Parameters:
map_to_organism (str or int) – organism name or NCBI taxon ID of the target species for ortholog mapping.
map_from_organism (str or int) – organism name or NCBI taxon ID of the input genes’ source species.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
filter_least_diverged (bool (default=True)) – if True (default), RNAlysis will only fetch ortholog mappings that were flagged as a ‘least diverged ortholog’ on the PantherDB database. You can read more about this flag on the PantherDB website: https://www.pantherdb.org/genes/
non_unique_mode ('first', 'last', 'random', or 'none' (default='first')) – How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
inplace (bool (default=True)) – If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- Returns:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- map_orthologs_phylomedb(map_to_organism: str | int | Literal, map_from_organism: Literal['auto'] | str | int | Literal = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', consistency_score_threshold: Fraction = 0.5, filter_consistency_score: bool = True, non_unique_mode: Literal['first', 'last', 'random', 'none'] = 'first', remove_unmapped_genes: bool = False, inplace: bool = True)
Map genes to their nearest orthologs in a different species using the PhylomeDB database. This function generates a table describing all matching discovered ortholog pairs (both unique and non-unique) and returns it, and can also translate the genes in this data table into their nearest ortholog, as well as remove unmapped genes.
- param map_to_organism:
organism name or NCBI taxon ID of the target species for ortholog mapping.
- type map_to_organism:
str or int
- param map_from_organism:
organism name or NCBI taxon ID of the input genes’ source species.
- type map_from_organism:
str or int
- param gene_id_type:
the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
- type gene_id_type:
str or ‘auto’ (default=’auto’)
- ` :param consistency_score_threshold: the minimum consistency score required for an ortholog mapping to be considered valid. Consistency scores are calculated by PhylomeDB and represent the confidence of the ortholog mapping. setting consistency_score_threshold to 0 will keep all mappings. You can read more about PhylomeDB consistency score on the PhylomeDB website: orthology.phylomedb.org/help
- type consistency_score_threshold:
float between 0 and 1 (default=0.5)
- param filter_consistency_score:
if True (default), when encountering non-unique ortholog mappings, RNAlysis will only keep the mappings with the highest consistency score.
- type filter_consistency_score:
bool (default=True)
- param non_unique_mode:
How to handle non-unique mappings. ‘first’ will keep the first mapping found for each gene; ‘last’ will keep the last; ‘random’ will keep a random mapping; and ‘none’ will discard all non-unique mappings.
- type non_unique_mode:
‘first’, ‘last’, ‘random’, or ‘none’ (default=’first’)
- param remove_unmapped_genes:
if True, rows with gene names/IDs that could not be mapped to an ortholog will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
- type remove_unmapped_genes:
bool (default=False)
- type inplace:
bool (default=True)
- param inplace:
If True (default), filtering will be applied to the current Filter object. If False, the function will return a new Filter instance and the current instance will not be affected.
- return:
DataFrame describing all discovered mappings (unique and otherwise). If inplace=True, returns a filtered instance of the Filter object as well.
- non_categorical_enrichment(background_genes: Set[str] | Filter | FeatureSet, attributes: List[str] | str | List[int] | int | Literal['all'], alpha: Fraction = 0.05, parametric_test: bool = False, attr_ref_path: str | Path | Literal['predefined'] = 'predefined', plot_log_scale: bool = True, plot_style: Literal['interleaved', 'overlap'] = 'overlap', n_bins: PositiveInt = 50, save_csv: bool = False, fname=None, return_fig: bool = False, gui_mode: bool = False) DataFrame | Tuple[DataFrame, List[Figure]]
Calculates enrichment and depletion of the FeatureSet for user-defined non-categorical attributes against a background set using either a one-sample T-test or Sign test. The attributes are drawn from an Attribute Reference Table. The background set is determined by either the input variable ‘background_genes’, or by the input variable ‘biotype’ and a Biotype Reference Table. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method).
- Parameters:
attributes (str, int, iterable (list, tuple, set, etc) of str/int, or 'all'.) – An iterable of attribute names or attribute numbers (according to their order in the Attribute Reference Table). If ‘all’, all of the attributes in the Attribute Reference Table will be used. If None, a manual input prompt will be raised.
alpha (float between 0 and 1 (default=0.05)) – Indicates the FDR threshold for significance.
parametric_test (bool (default=False)) – if True, performs a parametric statistical test (one-sample t-test). If False (default), performs a non-parametric statistical test (sign test).
background_genes (set of feature indices, filtering.Filter object, or enrichment.FeatureSet object) – a set of specific feature indices to be used as background genes.
attr_ref_path (str or pathlib.Path (default='predefined')) – the path of the Attribute Reference Table from which user-defined attributes will be drawn.
plot_log_scale (bool (default=True)) – if True (default), the Y-axis of the enrichment plot will be logarithmic. Otherwise, the Y-axis of the enrichment plot will be linear.
plot_style ('overlap' or 'interleaved' (default='overlap')) – ‘interleaved’ will plot an interleaved histogram. ‘overlap’ will plot a semi-transparent histogram where the obsreved and expected are overlapping.
n_bins (int larger than 0 (default=50)) – the number of bins to display in the enrichment plot histograms
save_csv (bool (default=False)) – If True, will save the results to a .csv file, under the name specified in ‘fname’.
fname (str or pathlib.Path (default=None)) – The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt.
return_fig (bool (default=False)) – if True, returns a matplotlib Figure object in addition to the results DataFrame.
- Return type:
pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure]
- Returns:
a polars DataFrame with the indicated attribute names in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
Example plot of non_categorical_enrichment(plot_style`=’overlap’)
Example plot of non_categorical_enrichment(plot_style=’interleaved’)
- pop()
Remove and return an arbitrary set element. Raises KeyError if the set is empty.
- ranked_genes
a vector of feature names/indices ordered by rank
- remove()
Remove an element from a set; it must be a member.
If the element is not a member, raise a KeyError.
- save_txt(fname: str | Path)
Save the list of features in the FeatureSet object under the specified filename and path.
- Parameters:
fname (str or pathlib.Path) – full filename/path for the output file. Can include the ‘.txt’ suffix but doesn’t have to.
- set_name
- single_set_enrichment(attributes: List[str] | str | List[int] | int | Literal['all'], alpha: Fraction = 0.05, min_positive_genes: PositiveInt = 10, lowest_cutoff: Fraction = 0.25, attr_ref_path: str | Path | Literal['predefined'] = 'predefined', exclude_unannotated_genes: bool = True, return_nonsignificant: bool = True, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False)
Calculates enrichment and depletion of the sorted RankedSet for user-defined attributes WITHOUT a background set, using the generalized Minimum Hypergeometric Test (XL-mHG, developed by Prof. Zohar Yakhini and colleagues and generalized by Florian Wagner). The attributes are drawn from an Attribute Reference Table. P-values are calculated using using the generalized Minimum Hypergeometric Test. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment = 0) appears with the smallest value in the scale.
- Parameters:
attributes (str, int, iterable (list, tuple, set, etc) of str/int, or 'all'.) – An iterable of attribute names or attribute numbers (according to their order in the Attribute Reference Table). If ‘all’, all of the attributes in the Attribute Reference Table will be used. If None, a manual input prompt will be raised.
alpha (float between 0 and 1) – Indicates the FDR threshold for significance.
attr_ref_path (str or pathlib.Path (default='predefined')) – path of the Attribute Reference Table from which user-defined attributes will be drawn.
exclude_unannotated_genes (bool (default=True)) – if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results.
return_nonsignificant (bool (default=True)) – if True (default), the results DataFrame will include all tested attributes - both significant and non-significant ones. If False, only significant attributes will be returned.
min_positive_genes (a positive int (default=10)) – the minimum number of ‘positive’ genes (genes that match the given attribute) for the enrichment to be considered a valid enrichment. All hypergeometric cutoffs with a smaller number of ‘positive’ genes will not be tested. This is the ‘X’ parameter of the XL-mHG nonparametric test. For example: a value of 10 means that a valid enrichment must have at least 10 ‘positive’ genes to be considered real enrichment.
lowest_cutoff (float between 0 and 1 (default=0.25)) – the lowest cutoff of the hypergeometric that will be tested. This determines the ‘L’ parameter of the XL-mHG nonparametric test. For example: a value of 1 means that every cutoff will be tested. A value of 0.25 means that every cutoff that compares the top 25% or smaller of the list to the rest of the list will be tested.
save_csv (bool, default False) – If True, will save the results to a .csv file, under the name specified in ‘fname’.
fname (str or pathlib.Path) – The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt.
return_fig (bool (default=False)) – if True, returns a matplotlib Figure object in addition to the results DataFrame.
plot_horizontal (bool (default=True)) – if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot.
show_expected (bool (default=False)) – if True, the observed/expected values will be shown on the plot.
plot_style ('bar' or 'lollipop' (default='bar')) – style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Return type:
pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure]
- Returns:
a polars DataFrame with the indicated attribute names in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
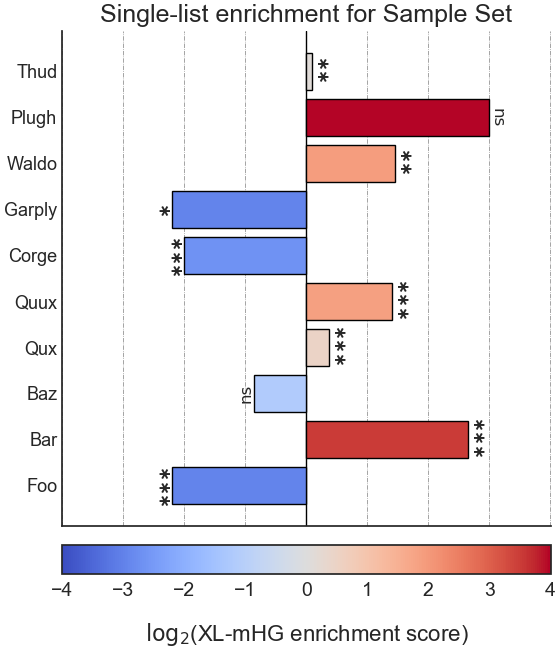
Example plot of single_set_enrichment()
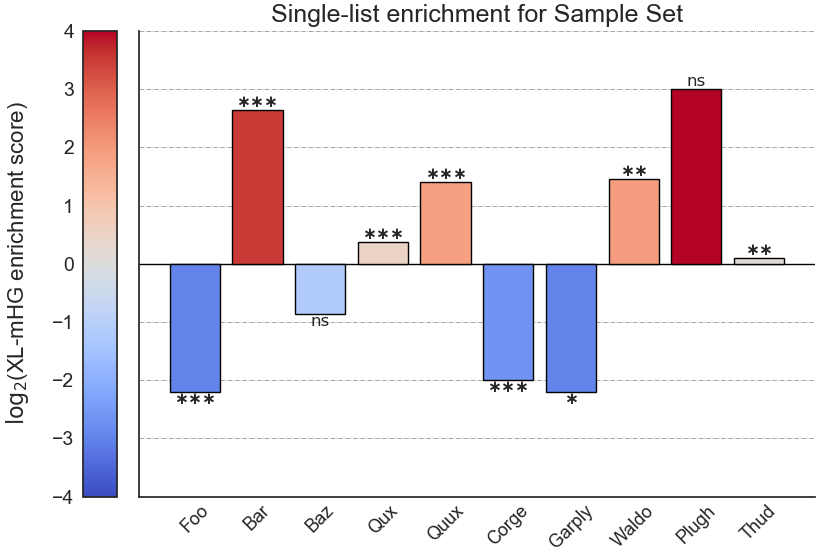
Example plot of single_set_enrichment(plot_horizontal = False)
- single_set_go_enrichment(organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', alpha: Fraction = 0.05, min_positive_genes: PositiveInt = 10, lowest_cutoff: Fraction = 0.25, propagate_annotations: Literal['classic', 'elim', 'weight', 'all.m', 'no'] = 'elim', aspects: Literal['any', 'biological_process', 'cellular_component', 'molecular_function'] | Iterable[Literal['biological_process', 'cellular_component', 'molecular_function']] = 'any', evidence_types: Literal['any', 'experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = 'any', excluded_evidence_types: Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic'] | Iterable[Literal['experimental', 'phylogenetic', 'computational', 'author', 'curator', 'electronic']] = (), databases: str | Iterable[str] | Literal['any'] = 'any', excluded_databases: str | Iterable[str] = (), qualifiers: Literal['any', 'not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'any', excluded_qualifiers: Literal['not', 'contributes_to', 'colocalizes_with'] | Iterable[Literal['not', 'contributes_to', 'colocalizes_with']] = 'not', exclude_unannotated_genes: bool = True, return_nonsignificant: bool = False, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', plot_ontology_graph: bool = True, ontology_graph_format: Literal['pdf', 'png', 'svg', 'none'] = 'none', parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) DataFrame | Tuple[DataFrame, Figure]
Calculates enrichment and depletion of the sorted RankedSet for Gene Ontology (GO) terms WITHOUT a background set, using the generalized Minimum Hypergeometric Test (XL-mHG, developed by Prof. Zohar Yakhini and colleagues and generalized by Florian Wagner). The GO terms and annotations are drawn via the GO Solr search engine GOlr, using the search terms defined by the user. P-values are calculated using the generalized Minimum Hypergeometric Test. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment = 0) appears with the smallest value in the scale.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID for which the function will fetch GO annotations.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the GOLR server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
alpha (float between 0 and 1) – Indicates the FDR threshold for significance.
min_positive_genes (a positive int (default=10)) – the minimum number of ‘positive’ genes (genes that match the given attribute) for the enrichment to be considered a valid enrichment. All hypergeometric cutoffs with a smaller number of ‘positive’ genes will not be tested. This is the ‘X’ parameter of the XL-mHG nonparametric test. For example: a value of 10 means that a valid enrichment must have at least 10 ‘positive’ genes to be considered real enrichment.
lowest_cutoff (float between 0 and 1 (default=0.25)) – the lowest cutoff of the hypergeometric that will be tested. This determines the ‘L’ parameter of the XL-mHG nonparametric test. For example: a value of 1 means that every cutoff will be tested. A value of 0.25 means that every cutoff that compares the top 25% or smaller of the list to the rest of the list will be tested.
propagate_annotations ('classic', 'elim', 'weight', 'all.m', or 'no' (default='elim')) – determines the propagation method of GO annotations. ‘no’ does not propagate annotations at all; ‘classic’ propagates all annotations up to the DAG tree’s root; ‘elim’ terminates propagation at nodes which show significant enrichment; ‘weight’ performs propagation in a weighted manner based on the significance of children nodes relatively to their parents; and ‘allm’ uses a combination of all proopagation methods. To read more about the propagation methods, see Alexa et al: https://pubmed.ncbi.nlm.nih.gov/16606683/
aspects (str, Iterable of str, 'biological_process', 'molecular_function', 'cellular_component', or 'any' (default='any')) – only annotations from the specified GO aspects will be included in the analysis. Legal aspects are ‘biological_process’ (P), ‘molecular_function’ (F), and ‘cellular_component’ (C).
evidence_types (str, Iterable of str, 'experimental', 'phylogenetic' ,'computational', 'author', 'curator', 'electronic', or 'any' (default='any')) – only annotations with the specified evidence types will be included in the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/
excluded_evidence_types (str, Iterable of str, 'experimental', 'phylogenetic' ,'computational', 'author', 'curator', 'electronic', or None (default=None)) – annotations with the specified evidence types will be excluded from the analysis. For a full list of legal evidence codes and evidence code categories see the GO Consortium website: http://geneontology.org/docs/guide-go-evidence-codes/
databases – only annotations from the specified databases will be included in the analysis. For a full list of legal databases see the GO Consortium website:
http://amigo.geneontology.org/xrefs :type databases: str, Iterable of str, or ‘any’ (default) :param excluded_databases: annotations from the specified databases will be excluded from the analysis. For a full list of legal databases see the GO Consortium website: http://amigo.geneontology.org/xrefs :type excluded_databases: str, Iterable of str, or None (default) :param qualifiers: only annotations with the speficied qualifiers will be included in the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type qualifiers: str, Iterable of str, or ‘any’ (default) :param excluded_qualifiers: annotations with the speficied qualifiers will be excluded from the analysis. Legal qualifiers are ‘not’, ‘contributes_to’, and/or ‘colocalizes_with’. :type excluded_qualifiers: str, Iterable of str, or None (default=’not’) :param exclude_unannotated_genes: if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results. :type exclude_unannotated_genes: bool (deafult=True) :param return_nonsignificant: if True, the results DataFrame will include all tested GO terms - both significant and non-significant terms. If False (default), only significant GO terms will be returned. :type return_nonsignificant: bool (default=False) :type save_csv: bool, default False :param save_csv: If True, will save the results to a .csv file, under the name specified in ‘fname’. :type fname: str or pathlib.Path :param fname: The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt. :type return_fig: bool (default=False) :param return_fig: if True, returns a matplotlib Figure object in addition to the results DataFrame. :type plot_horizontal: bool (default=True) :param plot_horizontal: if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot. :param show_expected: if True, the observed/expected values will be shown on the plot. :type show_expected: bool (default=False) :param plot_style: style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set. :type plot_style: ‘bar’ or ‘lollipop’ (default=’bar’) :type plot_ontology_graph: bool (default=True) :param plot_ontology_graph: if True, will generate an ontology graph depicting the significant GO terms and their parent nodes. :type ontology_graph_format: ‘pdf’, ‘png’, ‘svg’, or None (default=None) :param ontology_graph_format: if ontology_graph_format is not ‘none’, the ontology graph will additonally be generated in the specified file format. :type parallel_backend: Literal[PARALLEL_BACKENDS] (default=’loky’) :param parallel_backend: Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise. :rtype: pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure] :return: a polars DataFrame with the indicated attribute names in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
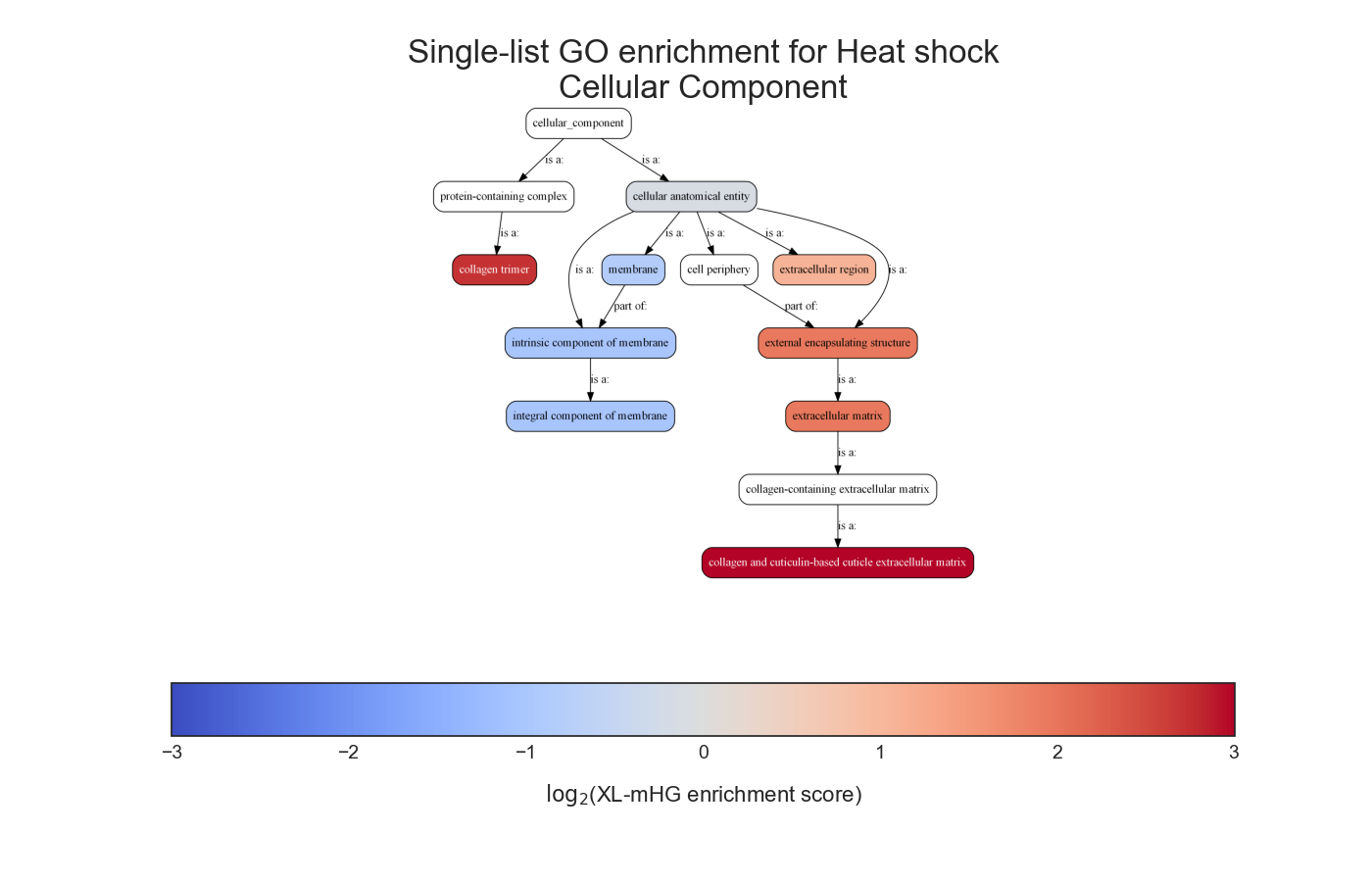
Example plot of single_set_go_enrichment(plot_ontology_graph=True)
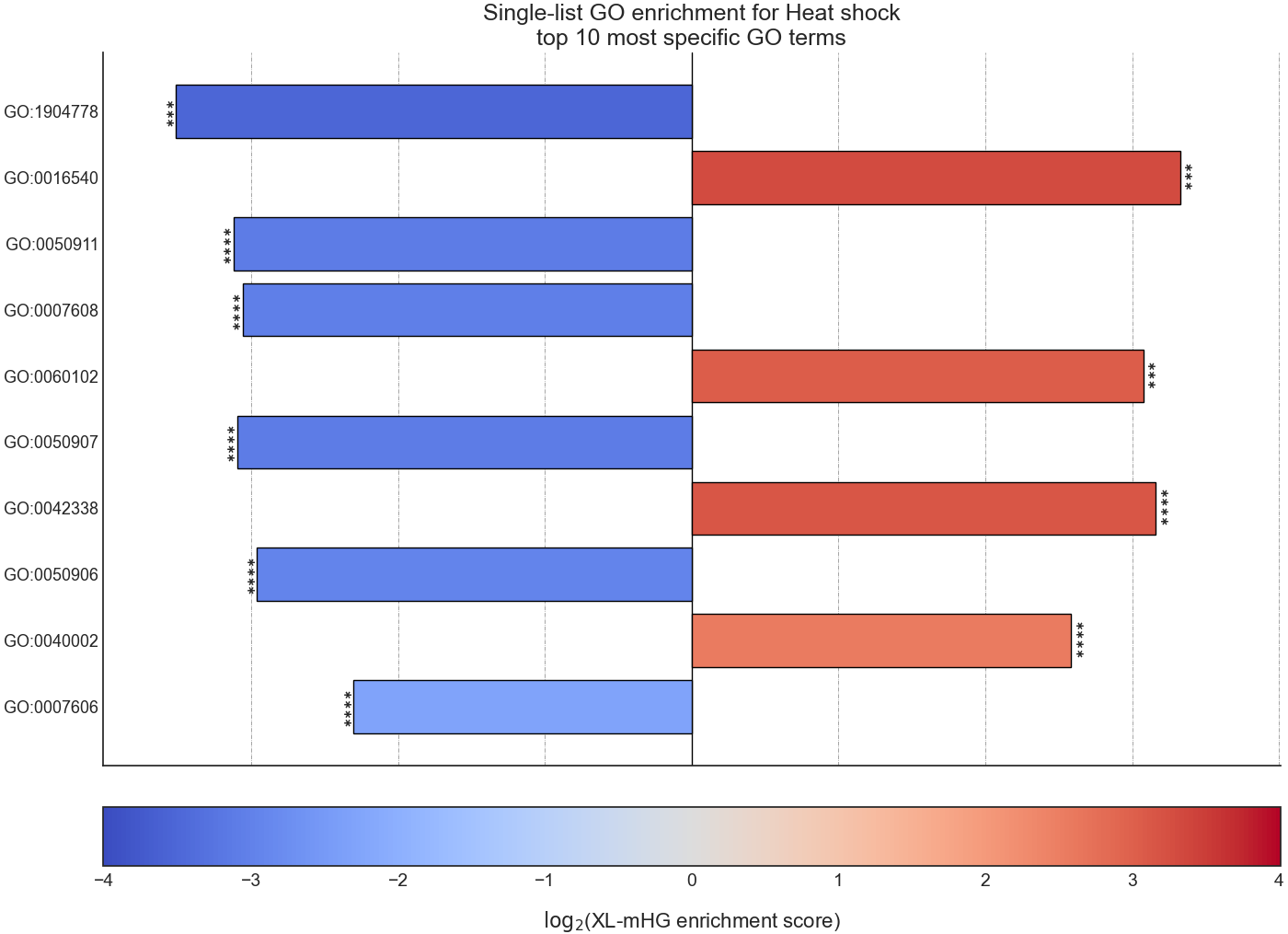
Example plot of single_set_go_enrichment()
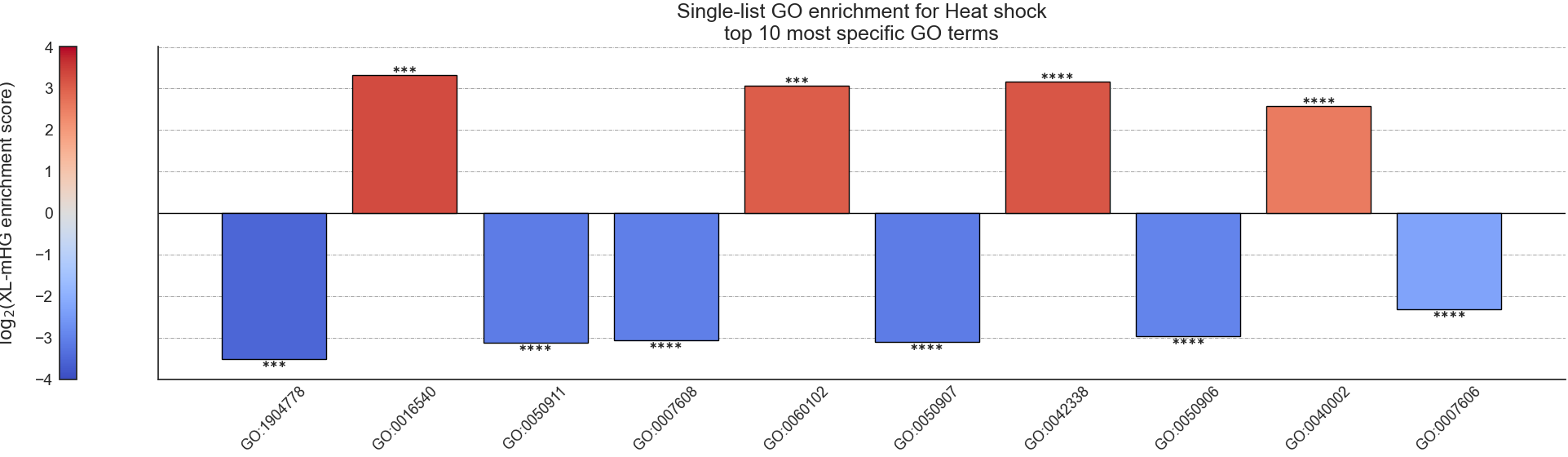
Example plot of single_set_go_enrichment(plot_horizontal = False)
- single_set_kegg_enrichment(organism: str | int | Literal['auto'] | Literal['Arabidopsis thaliana', 'Caenorhabditis elegans', 'Danio rerio', 'Drosophila melanogaster', 'Escherichia coli', 'Homo sapiens', 'Mus musculus', 'Saccharomyces cerevisiae', 'Schizosaccharomyces pombe'] = 'auto', gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', alpha: Fraction = 0.05, min_positive_genes: PositiveInt = 10, lowest_cutoff: Fraction = 0.25, exclude_unannotated_genes: bool = True, return_nonsignificant: bool = False, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', plot_pathway_graphs: bool = True, pathway_graphs_format: Literal['pdf', 'png', 'svg', 'none'] = 'none', parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) DataFrame | Tuple[DataFrame, Figure]
Calculates enrichment and depletion of the sorted RankedSet for Kyoto Encyclopedia of Genes and Genomes (KEGG) curated pathways WITHOUT a background set, using the generalized Minimum Hypergeometric Test (XL-mHG, developed by Prof. Zohar Yakhini and colleagues and generalized by Florian Wagner). P-values are calculated using the generalized Minimum Hypergeometric Test. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment = 0) appears with the smallest value in the scale.
- Parameters:
organism (str or int) – organism name or NCBI taxon ID for which the function will fetch GO annotations.
gene_id_type (str or 'auto' (default='auto')) – the identifier type of the genes/features in the FeatureSet object (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). If the annotations fetched from the KEGG server do not match your gene_id_type, RNAlysis will attempt to map the annotations’ gene IDs to your identifier type. For a full list of legal ‘gene_id_type’ names, see the UniProt website: https://www.uniprot.org/help/api_idmapping
alpha (float between 0 and 1) – Indicates the FDR threshold for significance.
min_positive_genes (a positive int (default=10)) – the minimum number of ‘positive’ genes (genes that match the given attribute) for the enrichment to be considered a valid enrichment. All hypergeometric cutoffs with a smaller number of ‘positive’ genes will not be tested. This is the ‘X’ parameter of the XL-mHG nonparametric test. For example: a value of 10 means that a valid enrichment must have at least 10 ‘positive’ genes to be considered real enrichment.
lowest_cutoff (float between 0 and 1 (default=0.25)) – the lowest cutoff of the hypergeometric that will be tested. This determines the ‘L’ parameter of the XL-mHG nonparametric test. For example: a value of 1 means that every cutoff will be tested. A value of 0.25 means that every cutoff that compares the top 25% or smaller of the list to the rest of the list will be tested.
min_positive_genes – the minimum number of ‘positive’ genes (genes that match the given attribute) for the enrichment to be considered a valid enrichment. All hypergeometric cutoffs with a smaller number of ‘positive’ genes will not be tested. This is the ‘X’ parameter of the XL-mHG nonparametric test. For example: a value of 10 means that a valid enrichment must have at least 10 ‘positive’ genes to be considered real enrichment.
lowest_cutoff – the lowest cutoff of the hypergeometric that will be tested. This determines the ‘L’ parameter of the XL-mHG nonparametric test. For example: a value of 1 means that every cutoff will be tested. A value of 0.25 means that every cutoff that compares the top 25% or smaller of the list to the rest of the list will be tested.
exclude_unannotated_genes (bool (deafult=True)) – if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results.
return_nonsignificant (bool (default=False)) – if True, the results DataFrame will include all tested GO terms - both significant and non-significant terms. If False (default), only significant KEGG pathways will be returned.
save_csv (bool, default False) – If True, will save the results to a .csv file, under the name specified in ‘fname’.
fname (str or pathlib.Path) – The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt.
return_fig (bool (default=False)) – if True, returns a matplotlib Figure object in addition to the results DataFrame.
plot_horizontal (bool (default=True)) – if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot.
show_expected (bool (default=False)) – if True, the observed/expected values will be shown on the plot.
plot_style ('bar' or 'lollipop' (default='bar')) – style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set.
plot_pathway_graphs (bool (default=True)) – if True, will generate pathway graphs depicting the significant KEGG pathways.
pathway_graphs_format ('pdf', 'png', 'svg', or None (default=None)) – if pathway_graphs_format is not ‘none’, the pathway graphs will additonally be generated in the specified file format.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Return type:
pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure]
- Returns:
a polars DataFrame with the indicated attribute names in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.

Example plot of single_set_kegg_enrichment(plot_pathway_graphs=True)
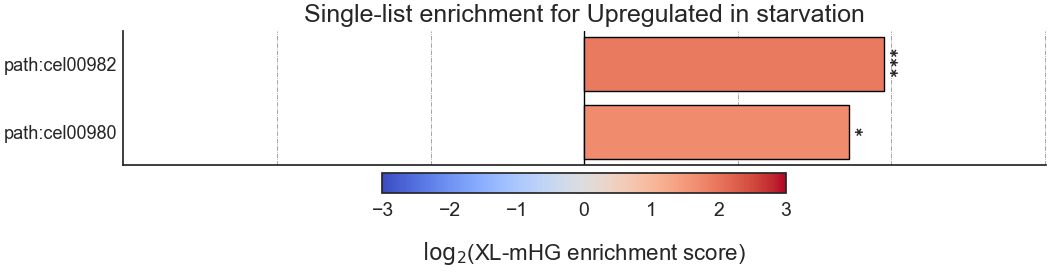
Example plot of single_set_kegg_enrichment()
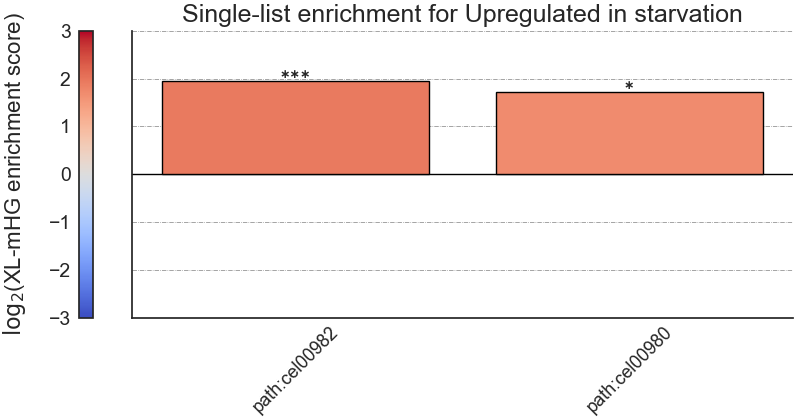
Example plot of single_set_kegg_enrichment(plot_horizontal = False)
- symmetric_difference(other: set | FeatureSet) FeatureSet
Calculates the set symmetric difference of the indices from two FeatureSet objects (the indices that appear in EXACTLY ONE of the FeatureSet objects, and not both/neither). A-symmetric difference-B is equivalent to (A-difference-B)-union-(B-difference-A).
- Parameters:
other (FeatureSet, RankedSet or set) – A second object against which the current object will be compared.
- Returns:
a new FeatureSet with elements in either this FeatureSet or the other object, but not both.
- Return type:
- Examples:
>>> from rnalysis import enrichment >>> en = enrichment.FeatureSet({'WBGene00000001','WBGene00000002','WBGene00000006'}, 'set name') >>> en2 = enrichment.FeatureSet({'WBGene00000004','WBGene00000001'}) >>> en.symmetric_difference(en2) >>> print(en) FeatureSet: set name {'WBGene00000002', 'WBGene00000006', 'WBGene00000004'}
- symmetric_difference_update()
Update a set with the symmetric difference of itself and another.
- translate_gene_ids(translate_to: str | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'], translate_from: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', remove_unmapped_genes: bool = False, inplace: bool = True) FeatureSet
Translates gene names/IDs from one type to another. Mapping is done using the UniProtKB Gene ID Mapping service. You can choose to optionally drop from the table all rows that failed to be translated.
- Parameters:
translate_to (str) – the gene ID type to translate gene names/IDs to. For example: UniProtKB, Ensembl, Wormbase.
translate_from (str or 'auto' (default='auto')) – the gene ID type to translate gene names/IDs from. For example: UniProtKB, Ensembl, Wormbase. If translate_from=’auto’, RNAlysis will attempt to automatically determine the gene ID type of the features in the table.
remove_unmapped_genes (bool (default=False)) – if True, rows with gene names/IDs that could not be translated will be dropped from the table. Otherwise, they will remain in the table with their original gene name/ID.
- Returns:
returns a new and translated FeatureSet.
- union(*others: set | FeatureSet) FeatureSet
Calculates the set union of the indices from multiple FeatureSet objects (the indices that exist in at least one of the FeatureSet objects).
- Parameters:
others (FeatureSet or set) – The objects against which the current object will be compared.
- Returns:
a new FeatureSet with elements from this FeatureSet and all other objects.
- Return type:
- Examples:
>>> from rnalysis import enrichment >>> en = enrichment.FeatureSet({'WBGene00000004','WBGene00000005','WBGene00000006'}, 'set name') >>> en2 = enrichment.FeatureSet({'WBGene00000004','WBGene00000001'}) >>> s = {'WBGene00000001','WBGene00000002','WBGene00000003'} >>> en.union(s, en2) >>> print(en) FeatureSet: set name {'WBGene00000003', 'WBGene00000004', 'WBGene00000001', 'WBGene00000002', 'WBGene00000006', 'WBGene00000005'}
- update()
Update a set with the union of itself and others.
- user_defined_enrichment(background_genes: Set[str] | Filter | FeatureSet, attributes: List[str] | str | List[int] | int | Literal['all'], statistical_test: Literal['fisher', 'hypergeometric', 'randomization'] = 'fisher', alpha: Fraction = 0.05, attr_ref_path: str | Path | Literal['predefined'] = 'predefined', exclude_unannotated_genes: bool = True, return_nonsignificant: bool = True, save_csv: bool = False, fname=None, return_fig: bool = False, plot_horizontal: bool = True, show_expected: bool = False, plot_style: Literal['bar', 'lollipop'] = 'bar', randomization_reps: PositiveInt = 10000, random_seed: int | None = None, parallel_backend: Literal['multiprocessing', 'loky', 'threading', 'sequential'] = 'loky', gui_mode: bool = False) DataFrame | Tuple[DataFrame, Figure]
Calculates enrichment and depletion of the FeatureSet for user-defined attributes against a background set. The attributes are drawn from an Attribute Reference Table. The background set is determined by either the input variable ‘background_genes’, or by the input variable ‘biotype’ and a Biotype Reference Table. P-values are corrected for multiple comparisons using the Benjamini–Hochberg step-up procedure (original FDR method). In plots, for the clarity of display, complete depletion (linear enrichment score = 0) appears with the smallest value in the scale.
- Parameters:
attributes (str, int, iterable (list, tuple, set, etc) of str/int, or 'all'.) – An iterable of attribute names or attribute numbers (according to their order in the Attribute Reference Table). If ‘all’, all of the attributes in the Attribute Reference Table will be used. If None, a manual input prompt will be raised.
statistical_test ('fisher', 'hypergeometric' or 'randomization' (default='fisher')) – determines the statistical test to be used for enrichment analysis. Note that some propagation methods support only some of the available statistical tests.
alpha (float between 0 and 1 (default=0.05)) – Indicates the FDR threshold for significance.
attr_ref_path (str or pathlib.Path (default='predefined')) – the path of the Attribute Reference Table from which user-defined attributes will be drawn.
biotype_ref_path (str or pathlib.Path (default='predefined')) – the path of the Biotype Reference Table. Will be used to generate background set if ‘biotype’ is specified.
biotype (str specifying a specific biotype, list/set of strings each specifying a biotype, or 'all' (default='protein_coding')) – determines the background genes by their biotype. Requires specifying a Biotype Reference Table. ‘all’ will include all genomic features in the reference table, ‘protein_coding’ will include only protein-coding genes from the reference table, etc. Cannot be specified together with ‘background_genes’.
background_genes (set of feature indices, filtering.Filter object, or enrichment.FeatureSet object) – a set of specific feature indices to be used as background genes.
exclude_unannotated_genes (bool (deafult=True)) – if True, genes that have no annotation associated with them will be excluded from the enrichment analysis. This is the recommended practice for enrichment analysis, since keeping unannotated genes in the analysis increases the chance of discovering spurious enrichment results.
return_nonsignificant (bool (default=True)) – if True (default), the results DataFrame will include all tested attributes - both significant and non-significant ones. If False, only significant attributes will be returned.
save_csv (bool (default=False)) – If True, will save the results to a .csv file, under the name specified in ‘fname’.
fname (str or pathlib.Path (default=None)) – The full path and name of the file to which to save the results. For example: ‘C:/dir/file’. No ‘.csv’ suffix is required. If None (default), fname will be requested in a manual prompt.
return_fig (bool (default=False)) – if True, returns a matplotlib Figure object in addition to the results DataFrame.
plot_horizontal (bool (default=True)) – if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot.
show_expected (bool (default=False)) – if True, the observed/expected values will be shown on the plot.
plot_style ('bar' or 'lollipop' (default='bar')) – style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set.
randomization_reps (int larger than 0 (default=10000)) – if using a randomization test, determine how many randomization repititions to run. Otherwise, this parameter will not affect the analysis.
parallel_backend (Literal[PARALLEL_BACKENDS] (default='loky')) – Determines the babckend used to run the analysis. if parallel_backend not ‘sequential’, will calculate the statistical tests using parallel processing. In most cases parallel processing will lead to shorter computation time, but does not affect the results of the analysis otherwise.
- Return type:
pl.DataFrame (default) or Tuple[pl.DataFrame, matplotlib.figure.Figure]
- Returns:
a polars DataFrame with the indicated attribute names in the first column; and a matplotlib Figure, if ‘return_figure’ is set to True.
Example plot of user_defined_enrichment()
Example plot of user_defined_enrichment(plot_horizontal = False)
- rnalysis.enrichment._fetch_sets(objs: dict, ref: str | Path | Literal['predefined'] = 'predefined')
Receives the ‘objs’ input from enrichment.upset_plot() and enrichment.venn_diagram(), and turns the values in it into python sets.
- Parameters:
objs (a dictionary, where the keys are names of sets, and the values are either python sets, FeatureSets or names of columns in the Attribute Reference Table.) – the ‘objs’ input given to the function enrichment.upset_plot() or enrichment.venn_diagram().
ref (str or pathlib.Path (default='predefined')) – the path of the Attribute Reference Table from which user-defined attributes will be drawn, if such attributes are included in ‘objs’.
- Returns:
a dictionary, where the keys are names of sets and the values are python sets of feature indices.
- rnalysis.enrichment.enrichment_bar_plot(results_table_path: str | Path, alpha: Fraction = 0.05, enrichment_score_column: str | Literal['log2_fold_enrichment', 'log2_enrichment_score'] = 'log2_fold_enrichment', n_bars: PositiveInt | Literal['all'] = 'all', title: str = 'Enrichment results', center_bars: bool = True, plot_horizontal: bool = True, ylabel: str | Literal['$\\log_2$(Fold Enrichment)', '$\\log_2$(Enrichment Score)'] = '$\\log_2$(Fold Enrichment)', ylim: float | Literal['auto'] = 'auto', plot_style: Literal['bar', 'lollipop'] = 'bar', show_expected: bool = False, title_fontsize: float = 18, label_fontsize: float = 13, ylabel_fontsize: float = 16) Figure
Generate an enrichment bar-plot based on an enrichment results table. For the clarity of display, complete depletion (linear enrichment = 0) appears with the smallest value in the scale.
- Parameters:
results_table_path (str or Path) – Path to the results table returned by enrichment functions.
alpha (float between 0 and 1 (default=0.05)) – the threshold for statistical significance. Used to draw significance asterisks on the graph.
enrichment_score_column (str (default='log2_fold_enrichment')) – name of the table column containing enrichment scores.
n_bars (int > 1 or 'all' (default='all')) – number of bars to display in the bar plot. If n_bars=’all’, all the results will be displayed on the graph. Otherwise, only the top n results will be displayed on the graph.
title (str) – plot title.
plot_horizontal (bool (default=True)) – if True, results will be plotted with a horizontal bar plot. Otherwise, results will be plotted with a vertical plot.
ylabel (str (default="$log_2$(Fold Enrichment)")) – plot y-axis label.
center_bars (bool (default=True)) – if True, center the bars around Y=0. Otherwise, ylim is determined by min/max values.
ylim (float or 'auto' (default='auto')) – set the Y-axis limits. If ylim`=’auto’, determines the axis limits automatically based on the data. If `ylim is a number, set the Y-axis limits to [-ylim, ylim].
plot_style ('bar' or 'lollipop' (default='bar')) – style for the plot. Either ‘bar’ for a bar plot or ‘lollipop’ for a lollipop plot in which the lollipop size indicates the size of the observed gene set.
show_expected (bool (default=False)) – if True, the observed/expected values will be shown on the plot.
title_fontsize (float (default=18)) – font size for the plot title.
label_fontsize (float (default=13)) – font size for the attribute labels on the plot.
ylabel_fontsize (float (default=16)) – font size for the y-axis colorbar label.
- Returns:
Figure object containing the bar plot
- Return type:
matplotlib.figure.Figure instance
- rnalysis.enrichment.gene_ontology_graph(aspect: Literal['biological_process', 'cellular_component', 'molecular_function'], results_table_path: str | Path, enrichment_score_column: str | Literal['log2_fold_enrichment', 'log2_enrichment_score'] = 'log2_fold_enrichment', title: str | Literal['auto'] = 'auto', ylabel: str = '$\\log_2$(Fold Enrichment)', graph_format: Literal['pdf', 'png', 'svg', 'none'] = 'none', dpi: PositiveInt = 300) Figure | None
Generate a GO enrichment ontology graph based on an enrichment results table.
- Parameters:
aspect ('biological_process', 'molecular_function', or 'cellular_component') – The GO aspect to generate an ontology graph for.
results_table_path (str or Path) – Path to the results table returned by enrichment functions.
enrichment_score_column (str (default='log2_fold_enrichment')) – name of the table column that contains the enrichment scores.
title (str or 'auto' (default='auto')) – plot title.
ylabel (str (default="$log_2$(Fold Enrichment)")) – plot y-axis label.
graph_format ('pdf', 'png', 'svg', or 'none' (default='none')) – if graph_format is not ‘none’, the ontology graph will additonally be generated in the specified file format.
dpi (int (default=300)) – resolution of the ontology graph in DPI (dots per inch).
- rnalysis.enrichment.kegg_pathway_graph(pathway_id: str, marked_genes: Sequence[str] | None, gene_id_type: str | Literal['auto'] | Literal['UniProtKB AC/ID', 'UniParc', 'UniRef50', 'UniRef90', 'UniRef100', 'Gene Name', 'CRC64', 'Ensembl', 'Ensembl Genomes', 'Ensembl Genomes Protein', 'Ensembl Genomes Transcript', 'Ensembl Protein', 'Ensembl Transcript', 'GeneID', 'KEGG', 'PATRIC', 'UCSC', 'WBParaSite', 'WBParaSite Transcript/Protein', 'ArachnoServer', 'Araport', 'CGD', 'ClinPGx', 'ConoServer', 'dictyBase', 'EchoBASE', 'euHCVdb', 'FlyBase', 'GeneCards', 'GeneReviews', 'HGNC', 'LegioList', 'Leproma', 'MaizeGDB', 'MGI', 'MIM', 'OpenTargets', 'Orphanet', 'PomBase', 'PseudoCAP', 'RGD', 'SGD', 'TubercuList', 'VEuPathDB', 'VGNC', 'WormBase', 'WormBase Protein', 'WormBase Transcript', 'Xenbase', 'ZFIN', 'eggNOG', 'GeneTree', 'HOGENOM', 'OMA', 'OrthoDB', 'CCDS', 'EMBL/GenBank/DDBJ', 'EMBL/GenBank/DDBJ CDS', 'GI number', 'PIR', 'RefSeq Nucleotide', 'RefSeq Protein', 'ChiTaRS', 'GeneWiki', 'GenomeRNAi', 'PHI-base', 'CollecTF', 'BioCyc', 'PlantReactome', 'Reactome', 'UniPathway', 'CPTAC', 'ProteomicsDB'] = 'auto', title: str | Literal['auto'] = 'auto', ylabel: str = '', graph_format: Literal['pdf', 'png', 'svg', 'none'] = 'none', dpi: PositiveInt = 300) Figure | None
Generate a KEGG Pathway graph.
- Parameters:
pathway_id (str) – KEGG ID of the pathway to be plotted.
marked_genes (sequence of str or None) – a set of genes/genomic features to be highlighted on the pathway graph. The gene ID type of those genes should match the parameter gene_id_type.
gene_id_type – the identifier type you want to use when displaying genes in the graph (for example: ‘UniProtKB’, ‘WormBase’, ‘RNACentral’, ‘Entrez Gene ID’). :type gene_id_type: str or ‘auto’ (default=’auto’)
title (str or 'auto' (default='auto')) – plot title.
ylabel (str (default="$log_2$(Fold Enrichment)")) – plot y-axis label.
graph_format ('pdf', 'png', 'svg', or 'none' (default='none')) – if graph_format is not ‘none’, the ontology graph will additonally be generated in the specified file format.
dpi (int (default=300)) – resolution of the ontology graph in DPI (dots per inch).
- rnalysis.enrichment.upset_plot(objs: Dict[str, str | FeatureSet | Set[str]], set_colors: ColorList = ('black',), title: str = 'UpSet Plot', title_fontsize: float = 20, show_percentages: bool = True, attr_ref_table_path: str | Path | Literal['predefined'] = 'predefined', fig: Figure = None) Figure
Generate an UpSet plot of 2 or more sets, FeatureSets or attributes from the Attribute Reference Table.
- Parameters:
objs (a dictionary with 2 or more entries, where the keys are the names of the sets, and the values are either a FeatureSet, a python set of feature indices, or a name of a column in the Attribute Reference Table. For example: {'first set':{'gene1','gene2','gene3'}, 'second set':'name_of_attribute_from_reference_table'}) – the FeatureSets, python sets or user-defined attributes to plot.
set_colors (Iterable of colors (default=('black',)) – If one color is supplied, this will determine the color of all sets on the plot. If multiple colors are supplied, this will determine the color of each set on the plot, and the subset colors will be determined by mixing.
title (str) – determines the title of the plot.
title_fontsize (float (default=20)) – font size for the plot’s title
show_percentages (bool (default=True)) – if True, shows the percentage that each set or subset takes out of the entire dataset.
attr_ref_table_path (str or pathlib.Path (default='predefined')) – the path of the Attribute Reference Table from which user-defined attributes will be drawn, if such attributes are included in ‘objs’.
fig (matplotlib.Figure) – optionally, supply your own Figure to generate the plot onto.
- Returns:
plt.Figure
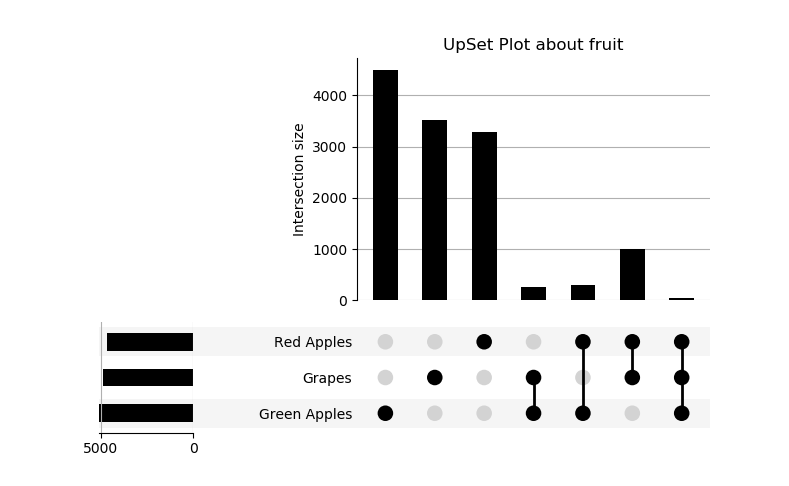
Example plot of upset_plot()
- rnalysis.enrichment.venn_diagram(objs: Dict[str, str | FeatureSet | Set[str]], title: str | Literal['default'] = 'default', attr_ref_table_path: str | Path | Literal['predefined'] = 'predefined', set_colors: ColorList = ('r', 'g', 'b'), transparency: Fraction = 0.4, weighted: bool = True, add_outline: bool = True, linecolor: Color = 'black', linestyle: Literal['solid', 'dashed'] = 'solid', linewidth: float = 2.0, title_fontsize: float = 14, set_fontsize: float = 12, subset_fontsize: float = 10, fig: Figure = None, **legacy_args) Figure
Generate a Venn diagram of 2 to 3 sets, FeatureSets or attributes from the Attribute Reference Table.
- Parameters:
objs (a dictionary with 2-3 entries, where the keys are the names of the sets, and the values are either a FeatureSet, a python set of feature indices, or a name of a column in the Attribute Reference Table. For example: {'first set':{'gene1','gene2','gene3'}, 'second set':'name_of_attribute_from_reference_table'}) – the FeatureSets, python sets or user-defined attributes to plot.
set_colors (tuple of matplotlib-format colors, the same size as 'objs') – determines the colors of the circles in the diagram.
attr_ref_table_path (str or pathlib.Path (default='predefined')) – the path of the Attribute Reference Table from which user-defined attributes will be drawn, if such attributes are included in ‘objs’.
title (str or 'default' (default='default')) – determines the title of the plot.
transparency (a float between 0 and 1) – determines the opacity of the circles. Opacity of 0 is completely transparent, while opacity of 1 is completely opaque.
weighted (bool (default=True)) – if True, the plot will be area-weighted.
add_outline (bool (default=True)) – if True, adds an outline to the circles.
linecolor (matplotlib-format color (default='black')) – Determines the color of the circles’ outline.
linestyle ('solid' or 'dashed' (default='solid')) – the style of the circles’ outline.
linewidth (float (default=2.0)) – the widdth of the circles’ outlines.
title_fontsize (float (default=14)) – font size for the plot’s title.
set_fontsize (float (default=12)) – font size for the set labels.
subset_fontsize (float (default=10)) – font size for the subset labels.
fig (matplotlib.Figure) – optionally, supply your own Figure to generate the plot onto.
- Returns:
a tuple of a VennDiagram object; and a list of 2-3 Circle patches.
Example plot of venn_diagram()
rnalysis.general module
This module contains general-purpose functions. Those include saving Filter objects and result tables, reading and updating the settings file, parsing common types of genomic feature indices, etc.
- rnalysis.general.parse_gene_name_string(string)
Receives a string that contains gene names (like ‘daf-2’ or ‘lin15B’). Parses the string into a set of gene names. The format of a gene name is a sequence consisting of the expression ‘[a-z]{3,4}’, the character ‘-’, and the expression ‘[A-Z,0-9]{1,4}’. :type string: str :param string: The string to be parsed. Can be any format of string. :return: a set of the WBGene indices that appear in the given string.
- Examples:
>>> from rnalysis import general >>> string = 'saeg-2 lin-15B cyp-23A1lin-15A WBGene12345678%GHF5H.3' >>> parsed = general.parse_gene_name_string(string) >>> print(parsed) {'saeg-2', 'lin-15B', 'cyp-23A1', 'lin-15A'}
- rnalysis.general.parse_sequence_name_string(string)
Receives a string that contains sequence names (such as ‘Y55D5A.5’). Parses the string into a set of WBGene indices. The format of a sequence name is a sequence consisting of the expression ‘[A-Z,0-9]{5,6}’, the character ‘.’, and a digit. :type string: str :param string: The string to be parsed. Can be any format of string. :return: a set of the WBGene indices that appear in the given string.
- Examples:
>>> from rnalysis import general >>> string = 'CELE_Y55D5A.5T23G5.6WBGene00000000 daf-16^^ZK662.4 ' >>> parsed = general.parse_sequence_name_string(string) >>> print(parsed) {'Y55D5A.5', 'T23G5.6', 'ZK662.4'}
- rnalysis.general.parse_wbgene_string(string)
Receives a string that contains WBGene indices. Parses the string into a set of WBGene indices. The format of a WBGene index is ‘WBGene’ and exactly 8 digits. :type string: str :param string: The string to be parsed. Can be any format of string. :return: a set of the WBGene indices that appear in the given string.
- Examples:
>>> from rnalysis import general >>> string = '''WBGene WBGenes WBGene12345678, WBGene98765432WBGene00000000& the geneWBGene44444444daf-16A5gHB.5 ... WBGene55555555''' >>> parsed = general.parse_wbgene_string(string) >>> print(parsed) {'WBGene12345678', 'WBGene44444444', 'WBGene98765432', 'WBGene55555555', 'WBGene00000000'}
- rnalysis.general.print_settings_file()
Print the current setting file configuration.
- Examples:
>>> from rnalysis import general >>> general.print_settings_file() Attribute Reference Table used: my_attribute_reference_table_path Biotype Reference Table used: my_biotype_reference_table_path
- rnalysis.general.reset_settings_file()
Resets the local settings by deleting the local settings file. Warning: this action is irreversible!
- rnalysis.general.save_to_csv(df: DataFrame | Filter, filename: str)
save a polars DataFrame or Filter object to csv. :type df: Filter object or polars DataFrame :param df: object to be saved :type filename: str :param filename: name for the saved file. Specify full path to control the directory where the file will be saved.
- rnalysis.general.set_attr_ref_table_path(path: str = None)
Defines/updates the Attribute Reference Table path in the settings file. :param path: the path you wish to set as the Attribute Reference Table path :type path: str
- Examples:
>>> from rnalysis import general >>> path="my_attribute_reference_table_path" >>> general.set_attr_ref_table_path(path) Attribute Reference Table path set as: my_attribute_reference_table_path
- rnalysis.general.set_biotype_ref_table_path(path: str = None)
Defines/updates the Biotype Reference Table path in the settings file. :param path: the path you wish to set as the Biotype Reference Table path :type path: str
- Examples:
>>> from rnalysis import general >>> path="my_biotype_reference_table_path" >>> general.set_biotype_ref_table_path(path) Biotype Reference Table path set as: my_biotype_reference_table_path
rnalysis.utils module
This module contains various utility functions. This module is used mainly by other modules, and is meant for internal use only.